3.5.1. Indels
Indel code is checked in to the csw-indel branch of [email protected]:santa-dev/santa-sim.git
There is a sample configuration file in examples/indel_test.xml
Configuration and specification of the indel model is Inspired by INDELible.
-- Fletcher, W. and Yang, Z. 2009. INDELible: a flexible simulator of biological sequence evolution. Mol. Biol. and Evol. 2009 26(8):1879-1888
Many virus species rely upon RNA polymerases or reverse-transcriptases for reproduction. These error-prone enzymes are a frequent source of mutations during replication that can have a significant impact on viral evolution. Errors introduced by polymerases include substitutions, random insertions or deletions, or stuttering and repeated motifs. These are not only a source of evolutionary diversity, but also can produce non-functional virions.
SANTA-sim was written to model viral evolution diversified by
substitutions and constrained by a broad palette of fitness functions.
Insertions and deletions (indels) in viral genomes were not originally supported
by SANTA-sim as doing so can be very difficult to simulate
efficiently.
We have added a prototype indel mutation model to SANTA-sim in order
to assess the impact on performance and to more closely mimic the
evolutionary behavior of retroviruses like HIV-1.
Extending SANTA to support indel mutations is disruptive to almost every assumption made by the program. Support for Indels breaks the strong asssumption that genome descriptions and the features defined on them are invariant.
In SANTA, each instance of class Genome is described by a
GenomeDescriptor which specifies the length and features defined
across the genome. A GenomeDescription is a relatively heavy-weighty
object, holding not only features coordinates, but also a one-to-one
mapping from genome-relative to feature-relative coordinates, for each
feature. In the absence of Indel mutations, there is only
one static GenomeDescriptor that serves to describe all genomes in a
population. For example, a GenomeDescription might specifiy the
genome ads having 2000 nucleotides with a single feature extending
from 300nt to 400nt. This is a suitable model in a system in which the
only mutations are substituions, but is insufficient to describe en
evolutionary model that includes indels.
Extending SANTA with indel mutations requires multiple
instances of GenomeDescription, one for each length and
combinations of features existing in a population. Instances of Genome were
extended with a reference to a shared instance of
GenomeDescription. Genomes with identical length and features,
descended from a common ancestor, will share a GenomeDescription.
There are several places in the SANTA code where an alignment of sequences in a population is assumed. Maintaining a global alignment of all genomes is not currently implemented so these
Indels are specified as part of the <nucleotideMutator> element in santa config files.
<mutator>
<nucleotideMutator>
<indelmodel model="NB">0.4 1</indelmodel>
<insertprob>2.5E-2</insertprob>
<deleteprob>2.5E-2</deleteprob>
</nucleotideMutator>
</mutator>
Indels are just another type of mutation, which along with substitutions, are the source of variability at each simulated replication of a parent genome. For each reproductive event, multiple base substitutions may occur but only a single insertion or deletion is allowed.
<insertprob> and <deleteprob> elements configure the probability
of an insertion or deletion event occurring per replication. These may
be set to the same values with <indelprob>. To ensure that only
zero or one indel occurs at each replication, a single value sampled
from an appropriately scaled uniform distribution is used to decide
between an insertion, deletion or no indel event, according to the
specified probabilities.1 If an indel occurs, the position of that
event is uniformly sampled from positions across the genome. If
unspecified, <insertprob> and <deleteprob> default to zero.
The model attribute in the <indelmodel> element specifies the distribution of indel lengths.
Currently only a Negative-Binomial distribution is supported, but additional length distributions are planned.
The length distribution is parameterized through values in the <indelmodel> element. In the example above, the Negative-Binomial distributions is supplied with two parameters,q=0.4 and r=1.
The length of indels is restricted to integer codon multiples to avoid throwing genomes out-of-frame. It is assumed that virtually all out-of-frame indels would result in non-productive transcripts. Avoiding production of those transcripts in the first place simplifies fitness calculations and speeds up the simulation.
[1] NucleotideMutator.java:124
In the nomenclature of SANTA config files, <feature> elements define regions of a genome roughly corresponding to a gene.
Features, along with factors, anchor fitness functions to specific regions of the genome.
When the only mutations are substitutions, features never move and once defined on founder genomes, are also valid on all descendants.
Fitness factors define sub-regions of features over which specific scoring functions are applied.
Examples of fitness factors are <purifyingFitness>, <empiricalFitness>, and <populationSizeDependentFitness>.
Factors are defined with feature-relative coordinates in units of
nucleotides or amino acids, as specified by the <type> definition of
the Feature.
Shown below is an example of some feature and factor definitions.
<genome>
<feature>
<name>PR</name>
<type>aminoAcid</type>
<coordinates>27-116</coordinates>
</feature>
<feature>
<name>CDS</name>
<type>aminoAcid</type>
<coordinates>1-609</coordinates>
</feature>
</genome>
<fitnessFunction>
<purifyingFitness>
<feature>CDS</feature>
<sites>1-203</sites>
<fitness>
<lowFitness>0.9</lowFitness>
<minimumFitness>0.1</minimumFitness>
</fitness>
</purifyingFitness>
<populationSizeDependentFitness>
<feature>PR</feature>
<sites>1-30</sites>
<declineRate>0.005</declineRate>
<maxPopulationSize>50000</maxPopulationSize>
</populationSizeDependentFitness>
</fitnessFunction>
Indel mutations invalidate the assumption of a static genome length. Clearly if nucleotides are being inserted or deleted in the genome, the features defined on that genome will have to shift or change size in response. The challenge in adding indel support to SANTA-sim was defining exactly how feature and fitness factors respond as indels change the underlying nucleotide sequence.
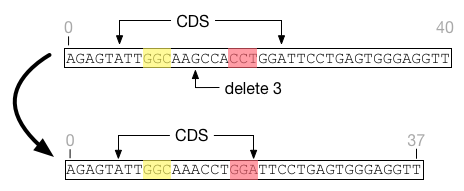
Of critical importance is determining where an indel begins with
respect to a feature. If we picture the genome laid out
left-to-right, insertions to the left of a feature will shift that
feature to the right without affecting its size. Similarly deletions
that occur entirely to the left of feature will shift the feature to
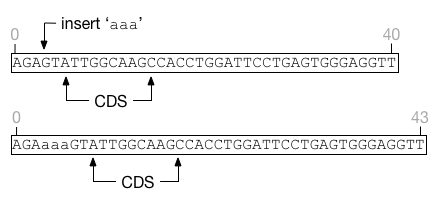
the left. For example, in this figure the feature CDS is shifted
to the right by an insertion. The feature covers exactly the same nucleotides
before and after the insertion event.
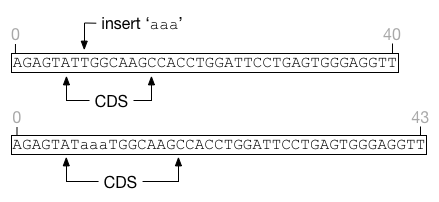
Insertions that land in the middle of a feature will extend the feature length without affecting its start position. Deletions entirely within a feature will shrink the feature without affecting its start position. It is possible for a feature to shrink to nothing, in which case it cannot be resurrected regardless of insertion events that may occur in the future.
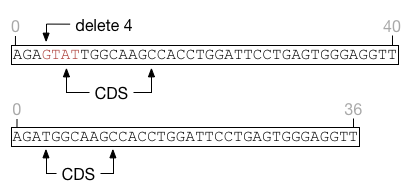
Insertions immediately to the left or right of a feature do not extend the feature and can only shift the feature on one direction or another.
Deletions overlapping the start or end of a feature are more complicated as they can simultaneously affect a feature's size and position, as in this example.
Some features consist of multiple fragments, e.g.
<feature>
<name>Example</name>
<coordinates>10-35,100-150</coordinates>
</feature>
The rules for adjusting features apply to each individual fragment as well.
As described above, factors are regions defined on features over which fitness scoring functions are applied.
For example, the following configuration defines a factor that applies a purifying selection fitness function over a region of the CDS feature from the first nucleotide to the ninth. The <sites> coordinates are inclusive 1-based nucleotide positions relative to the start of the feature.
<fitnessFunction>
<purifyingFitness>
<feature>CDS</feature>
<sites>1-9</sites>
<rank>
<order>observed</order>
<probableSet>2</probableSet>
<breakTies>random</breakTies>
</rank>
<fitness>
<lowFitness>0.1</lowFitness>
<minimumFitness>0.1</minimumFitness>
</fitness>
</purifyingFitness>
</fitnessFunction>
Factor coordinates are not affected by indels. They are defined by feature-relative coordinates and do not need to change as long as the underlying feature coordinates are adjusted. For example, the highlighted region in the figure below shows how factor coordinates respond as the underlying feature is affected by indels. At all times the factor is defined as beginning at the third position in the feature and ending at the sixth. Although the underlying features shift and change size, the factor does not.
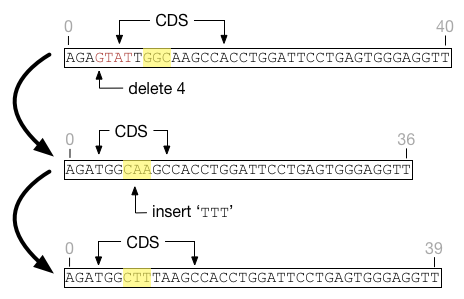
Indels may cause a feature to shrink such that factors fall outside the boundaries of that feature. Fitness functions are not defined on sites outside the boundaries of their underlying feature, and such sites will be silently ignored. If the feature grows again to encompass the factor, those sites will again be well-defined.
For each insertion, deletion or substitution on a genome, there is a corresponding change in fitness. SANTA-sim tracks fitness changes on a per-site basis. That is, a substitution at a site will cause a record to be created that notes the position, the previous base at the site, and the new base at the site. In this way the fitness score for that genome can be quickly updated without reevaluating fitness across the entire genome. Substitutions are relatively straight-forward to implement in this scheme, but indels again present challenges for this fitness model.
A primary challenge is that indels can have fitness repercussions across multiple sites, even beyond the boundaries of the indel itself. In the figure below, two factors are represented by the yellow and red highlighted regions. After a deletion, the feature shrinks and the content bounded by the red factor changes. Thus a region seemingly disconnected from the indel event requires a change in fitness score.
The new indel feature does not try to be clever in updating fitness functions. Whenever a deletion or insertion event occurs, the fitness cache for the genome is deleted and the fitness value is recalculated across the entire genome.
SANTA implements a simplified model of recombination capable of simulating transitions between two templates of single-segment viruses. The probability of recombination at any generation is a product of the probability of multiple infection of a single cell and the probability of a template switch during replication. The number and position of transitions are both chosen from random distributions. No attempt is made to identify regions of homology between the two genomes.
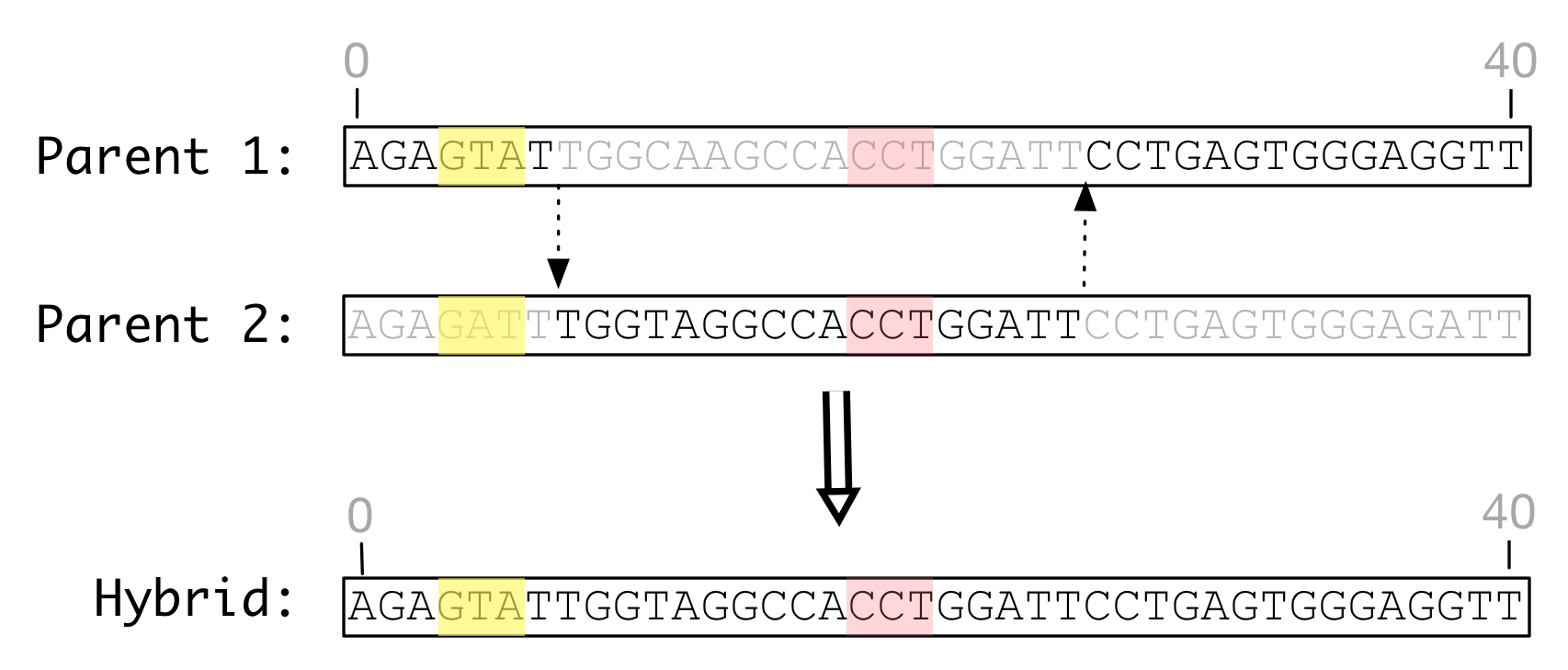
In the absence of indel mutations, all genomes are of identical length and recombination is a relatively simple task deciding the number and positions of transitions and concatenating the parent fragments into a complete hybrid. A simplifying assumption is that template switching occurs synchronously; moving ahead or back when switching templates is not allowed. The resulting hybrid genome has exactly the same length and features as its parents. (Recall that features roughly correspond to genes in a real genome.)
Shown below is a simulated recombination between two genome of identical length that have undergone only substitution mutation. Each features identical feature definitions illustrated by colored regions.
With the introduction of simulated indel mutations, co-infecting genomes may be of different lengths, and resolving a single set of feature definitions in the hybrid product requires careful navigation of the feature set in each parent. Depending on the evolutionary history of the parents, recombination may partially or wholly duplicate or eliminate features.
Shown below is an illustration of a double recombination event between two templates that have undergone substitution and indel mutations. The recombined product shows the paralogous duplication of one feature and the complete elimination of another.
 Figure Recombination with indels: Indel
mutations may result in recombining parent genomes of different lengths
and feature definitions. The hybrid product is likely to have
a different length than either parent, and may modify, duplicate, or
eliminate some feature definitions.
Figure Recombination with indels: Indel
mutations may result in recombining parent genomes of different lengths
and feature definitions. The hybrid product is likely to have
a different length than either parent, and may modify, duplicate, or
eliminate some feature definitions.
As described above, simulated recombination is not homology-driven. Unlike in-vivo recombination, transition points are selected randomly without regard for nucleotide composition. Viral- or cellular-recombination can be more accurately modelled by distributing transition points in a homology-aware manner. Homologous regions can be identified by performing a global alignment of the parent genomes before recombination. The recombined product may have a different length than either parent, and feature definitions may change length, be duplicated, or disappear altogether.
The figure below shows homologous recombination between two genomes of identical length that have been mutated by substitutions but not indels. The parent genomes are aligned with one another to identify homologous sites in each genome. When recombined, the hybrid product has a different length than either parent and one feature has shifted position.
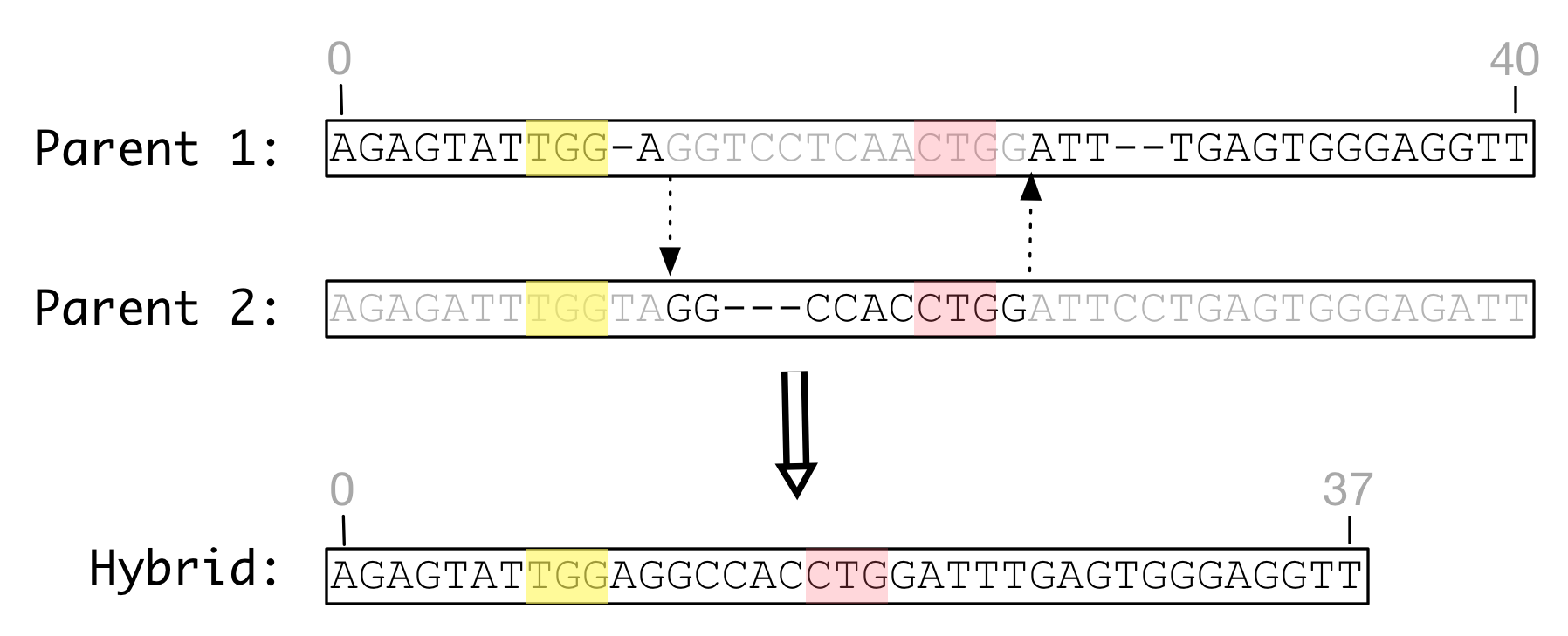 Figure Homologous recombination: Homologous recombination of
identical length genomes with identical feature definitions
can produce a hybrid of a different length with modified feature
definitions.
Figure Homologous recombination: Homologous recombination of
identical length genomes with identical feature definitions
can produce a hybrid of a different length with modified feature
definitions.
Unlike homology-naive recombination, the distribution of break points must be aware of the content in each aligned parent to avoid choosing positions at which one of the parents has a gap. Homologous recombination requires pairwise global alignment which may be computationally expensive, but is only necessary after the (presumably rare) decision to undergo recombination has been made.
Homologous recombination is not currently implemented in SANTA.
- Indels are only implemented for derivatives of the
SimpleGenomeclass.CompactGenomedoes not yet have indel support. - Distance calculations are not implemented among genomes of different lengths.
- Calculation of consensus sequences among genomes of different lengths is not yet supported.