Magellan is an R package to identify locations within a spatial transcriptomics experiment that are undergoing a biological response to a perturbation.
Magellan builds on our Augur package for cell type prioritization within single-cell transcriptomics data. The intuition underlying Augur is that cells undergoing a profound response to a given experimental stimulus become more separable, in the space of molecular measurements, than cells that remain unaffected by the stimulus. Augur quantifies this separability by asking how readily the experimental sample labels associated with each cell (e.g., treatment vs. control) can be predicted from molecular measurements alone. This is achieved by training a machine-learning model specific to each cell type, to predict the experimental condition from which each individual cell originated. The accuracy of each cell type-specific classifier is evaluated in cross-validation, providing a quantitative basis for cell type prioritization.
Magellan extends this concept by training a machine-learning model specific to a small two-dimensional region within a spatial transcriptomics experiment, centred on a particular barcode. Instead of retrieving cells of a particular type as input to the classifier, Magellan retrieves a set of neighboring barcodes, within the coordinate system of the tissue of interest. Magellan then computes the accuracy with which the experimental condition can be predicted for the spatial nearest-neigbors of each barcode in turn. This approach allows Magellan to circumscribe the perturbation response to specific regions of a complex tissue.
Magellan relies on functions from the following R packages:
Augur,
dplyr (>= 0.8.0),
purrr (>= 0.3.2),
tibble (>= 2.1.3),
magrittr (>= 1.5),
tester (>= 0.1.7),
Matrix (>= 1.2-14),
sparseMatrixStats (>= 0.1.0),
parsnip (>= 0.0.2),
recipes (>= 0.1.4),
rsample (>= 0.0.4),
yardstick (>= 0.0.3),
pbmcapply (>= 1.5.0),
lmtest (>= 0.9-37),
randomForest (>= 4.6-14),
tidyselect (>= 0.2.5),
rlang (>= 0.4.0),
tidyr (>= 1.1.2),
RANN (>= 2.6.1)
In addition, the Seurat package must be installed for Magellan to take a Seurat object as input.
Magellan has been tested with R version 4.1.0 and higher.
To install Magellan, first install the devtools package, if it is not already installed:
> install.packages("devtools")Then, install the sparseMatrixStats package from GitHub using devtools:
> devtools::install_github("const-ae/sparseMatrixStats")Finally, install Augur and Magellan from GitHub:
> devtools::install_github("neurorestore/Augur")
> devtools::install_github("neurorestore/Magellan")This should take no more than a few minutes.
The main function of Magellan, navigate_space, takes three pieces of information as input:
input: a processed gene expression matrix, with genes in rows and spatial barcodes in columnsmeta: a data frame containing metadata associated with each barcode, minimally including the experimental condition from which each barcode was obtainedcoords: the two-dimensional coordinates of each barcode within the tissue section
Optionally, all three can all be provided jointly as input in the form of a Seurat object.
To run Augur with default parameters on a spatial transcriptome scRNA-seq matrix expr, and accompanying data frames meta and coords, call the navigate_space function like so:
> results = navigate_space(expr, meta, coords)Magellan assumes that the experimental conditions are provided in the 'label' column of the meta data frame, and coordinates are provided in the 'coord_x' and 'coord_y' columns of the coords data frame. To change the names of these columns, specify the label_col and coord_cols arguments. For example, to run Magellan on a dataset where the experimental conditions are in the column 'group' and the spatial coordinates are in columns 'xpos' and 'ypos', call navigate_space like so:
> results = navigate_space(expr, meta, coords, label_col = 'group', coord_cols = c('xpos', 'ypos'))Spatial prioritizations take the form of an AUC for each barcode, and are stored in the AUC data frame returned by Magellan:
> head(results$AUC, 5)
# A tibble: 6 x 2
barcode auc
<chr> <dbl>
1 Cell68_1 0.625
2 Cell129_1 0.578
3 Cell43_1 0.550
4 Cell14_1 0.617
5 Cell51_1 0.602
6 Cell85_1 0.601
... ...Magellan can also run directly on a Seurat object. For a Seurat object sc containing metadata and spatial coordinates, simply do:
> results = navigate_space(sc)To see Magellan in action, load the simulated spatial transcriptomics dataset that is bundled with the Magellan package:
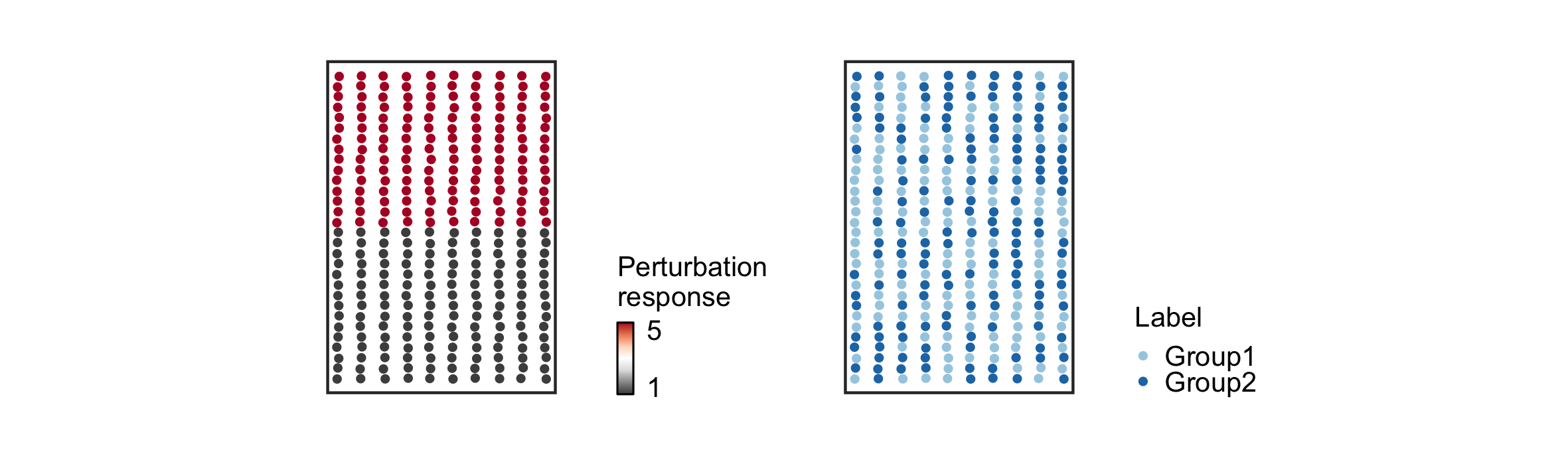
> data("demo")This toy dataset consists of 300 spatial barcodes, distributed approximately evenly across a two-dimensional tissue in a grid-like pattern, with 150 barcodes sampled from each of two biological conditions. The tissue consists of two regions, one on the top and one on the bottom, that are undergoing distinct responses to a biological perturbation. Specifically, the top region is undergoing a more profound transcriptional response to the perturbation of interest.
We can run Magellan on this dataset, which is provided as a Seurat object, using the following code. This should take about 30 minutes. Note that for the sake of runtime, here we use only 5 iterations of cross-validation per barcode. This leads to an increased amount of noise in the results (which would be mitigated by using the default setting of 50 iterations), at the cost of an increased runtime. Please also note that by default, Magellan will run on 32 threads; decrease this number by specifying the n_threads argument to navigate_space.
> results = navigate_space(demo, n_subsamples = 5)We can inspect the spatial prioritization by extracting the AUCs for each barcode and overlaying them onto the two-dimensional coordinates of the image:
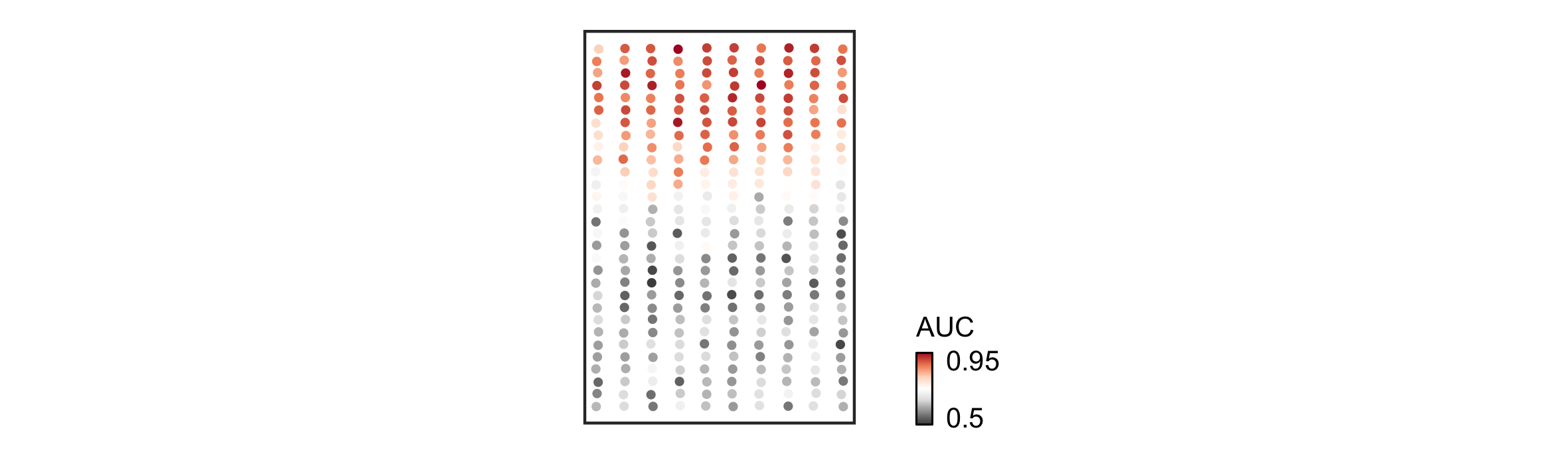
Magellan has correctly recovered the simulated spatial pattern of perturbation responsiveness.