Diversity and composition
- Analysis of diversity and composition
Alpha diversity represents within-sample diversity. We can now make use of the Shannon diversity metric information we added to our phyloseq objects earlier to investigate whether there are changes based on some sample metadata of our choosing.
Alpha diversity measures and an introduction to Hill diversity indices are summarised nicely in the article "A conceptual guide to measuring species diversity".
- Chao1 diversity is a measure of richness, and even the best asymptotic richness estimators (such as Chao1) cannot reliably predict the true community diversity. The problem lies in that richness measurements are strongly influenced by the rarest species, which are precisely the species that we know least about. Another way to put this is that richness has a high uncertainty.
- The "traditional" diversity indices that explicitly include relative abundance, such as the Shannon and Simpson indices, are more robust than richness to the sampling problems that affect richness. However, these measures have their own units and do not scale intuitively (or even similarly) with species gain and loss.
- Hill–Shannon diversity lies between richness and Hill–Simpson diversity, and may be the ‘just right' measure in many applications (Kempton 1979). The geometric mean affords leverage to extreme values according to their proportional, not absolute, difference from the mean. Thus, it can respond strongly to both very high and to very low rarity values. Another argument in favor of Hill–Shannon is that many species abundance distributions are approximately log-normal (Williamson and Gaston 2005, McGill et al. 2007), and thus their central tendency might be well described by the geometric mean.
The approach for this will be different depending on whether your metadata of interest is categorical or numerical, and how you want to split up your data (if at all – for example, you may have a numeric explanatory variable, but also a categorical grouping factor).
Perhaps we have a longitudinal study where faecal samples have been taken from the same animals at five distinct time points, and the animals belong to one of two treatment groups, e.g. a Sham group and a drug treatment group.
Firstly we may want to determine if there are changes from the baseline alpha diversity (timepoint 0). One way we could investigate that is as follows:
# Transform the sample data into a data.frame
bact_data_samples <- data.frame(sample_data(bact_data_filtered))
# Plot the diversity over time by group
(bact_diversity_plot <- ggplot(bact_data_samples, aes(x = days_postTx, y = Diversity)) +
geom_path(aes(group = rat_id), col = 'grey70') + # draw a path between sample from the same animal
geom_violin(aes(y = Diversity, fill = group), scale = 'area') +
geom_dotplot(binaxis = 'y', stackdir = 'center', dotsize = 0.5, binwidth = 1/30 * diff(range(bact_data_samples$Diversity))) +
geom_boxplot(aes(y = Diversity, fill = group), width = 0.2) +
scale_fill_jama(alpha = 0.5, name = 'Treatment Group') +
guides(fill = 'none') +
labs(title = 'Bacterial Diversity (Shannon Index)',
x = 'Time Since Treatment (days)') +
facet_grid(cols = vars(group)) +
stat_compare_means(comparisons = list(c('0', '7'),
c('0', '14'),
c('0', '21'),
c('0', '28')))
)
ggsave(here::here('figures', 'diversity', 'bact_diversity_by_group.pdf'), bact_diversity_plot,
width = 16, height = 12, units = 'cm')That would produce this plot:

Alternatively, we may decide we want to look at whether the treatment affects alpha diversity between groups over time. For example, there may be some temporal element affecting alpha diversity that is independent of the treatment. This is easy to alter by changing the faceting variable, and removing the geom_path() element.
# Transform the sample data into a data.frame
bact_data_samples <- data.frame(sample_data(bact_data_filtered))
# Prepare facet labels (more descriptive than the numbers alone)
# We will use the labeller within facet_grid to use these labels
facet_col_labels <- c('0' = '0 days post-Tx',
'7' = '7 days post-Tx',
'14' = '14 days post-Tx',
'21' = '21 days post-Tx',
'28' = '28 days post-Tx')
# Plot the diversity over time between groups
(bact_diversity_plot2 <- ggplot(bact_data_samples, aes(x = group, y = Diversity)) +
geom_violin(aes(y = Diversity, fill = group), scale = 'area') +
geom_dotplot(binaxis = 'y', stackdir = 'center', dotsize = 0.5,
binwidth = 1/30 * diff(range(bact_data_samples$Diversity))) +
geom_boxplot(aes(y = Diversity, fill = group), width = 0.2) +
scale_fill_jama(alpha = 0.5, name = 'Treatment Group') +
guides(fill = 'none') +
labs(title = 'Bacterial Diversity (Shannon Index)',
x = 'Treatment Group') +
facet_grid(cols = vars(days_postTx), scales = 'free', labeller = as_labeller(facet_col_labels)) +
stat_compare_means(comparisons = list(c('Sham', 'DrugXYZ')))
)
ggsave(here::here('figures', 'diversity', 'bact_diversity_by_time.pdf'), bact_diversity_plot2,
width = 16, height = 12, units = 'cm')That would produce this plot:

We can also loop through all the available diversity metrics and produce a combined plot with ggarrange as follows:
# Plot the diversity metrics
diversity_plots <- list()
for (i in c('Chao1', 'Shannon', 'Simpson', 'Hill_Shannon_phylo', 'Hill_Shannon_taxa')) {
p <- ggplot(bact_df_samples, aes(x = group, y = .data[[i]])) +
geom_violin(aes(y = .data[[i]], fill = group), scale = 'area', trim = FALSE) +
geom_boxplot(aes(y = .data[[i]], fill = group), width = 0.2) +
geom_dotplot(binaxis = 'y', stackdir = 'center', dotsize = 1) +
scale_fill_jama(alpha = 0.5, name = 'Group') +
scale_color_aaas() +
labs(title = paste0(i, ' Index'),
x = 'Group',
y = paste0(i, ' Diversity Index')) +
stat_compare_means(comparisons = list(c('Sham', 'DrugXYZ')), size = 3) +
theme_pubr(legend = 'right')
diversity_plots[[i]] <- p
}
p_arr <- ggarrange(plotlist = diversity_plots, nrow = 1, common.legend = TRUE, legend = 'none')
ggsave(here::here('figures', 'diversity', 'treatment_diversity_metrics.pdf'), p_arr,
width = 35, height = 8, units = 'cm')Beta diversity represents between-sample diversity. Continuing on from our example above, because the taxonomic composition may vary greatly with time, if we were to plot ordination plots for all samples together, we may lose clarity and minimise resolution/separation of data points at each of the time points individually.
In this example, the data came from 16S rRNA amplicon sequencing, so we have access to the de novo phylogenetic tree, and can use the UniFrac distance metric. UniFrac distances make use of the tree, by assigning short distances to a pair of ASVs that are highly similar, and larger distances to those that are more dissimilar.
If however we were using a phyloseq derived from shotgun metagenomic sequencing data, we could use another distance metric, e.g. Bray-Curtis ('bray').
To save time and code, let's loop through the ordinations at each timepoint by creating temporary phyloseq subsets, add their plotted ordinations to a plot list, and finally combine the plots into a single figure with ggarrange().
If you just have a single categorical variable however, you can make use of the plot code within the loop, i.e. p <- plot_ordination() etc.
# Choose parameters for downstream analyses
met <- 'PCoA'
dist <- 'unifrac'
bact <- bact_data_logCSS
# Bacterial ordination (separately for each temporal group)
ord_list <- list() # blank list to hold ordination plots
for (day in levels(bact@sam_data$days_postTx)) { # loop through the unique values of 'days_postTx' in the phyloseq sample_data()
bact_tmp <- prune_samples(bact@sam_data$days_postTx == day, bact) # create temporary subsets of the phyloseq object
p <- plot_ordination(bact_tmp, ordinate(bact_tmp, met, dist, weighted = TRUE), # ordinate via the phyloseq plot_ordination() function
title = paste0(day, ' days post-treatment')) +
stat_ellipse(aes(fill = group), geom = 'polygon', type = 't', level = 0.95, alpha = 0.2) + # add group ellipses
scale_shape_identity() +
geom_point(aes(fill = group), shape = 21, size = 3) + # add the individual data points
scale_fill_jama(name = 'Treatment Group') # scale the fill colour using ggsci scale_fill_jama() function
ord_list[[day]] <- p # add the plot to the plot list
}
# Arrange ordination plots using ggpubr ggarrange() function
ord_plots <- ggarrange(plotlist = ord_list, nrow = 2, ncol = 3, common.legend = TRUE)
(ord_plots <- annotate_figure(ord_plots,
top = text_grob(label = paste0('Bacteria ', met, ' ', dist, ' ordination'))))
ggsave(here::here('figures', 'ordination', 'bact_PCoA_ordination_by_time.pdf'), ord_plots, # Save combined figure
width = 20, height = 16, units = 'cm')The resulting output file looks like this:

In addition to this, performing a PERMANOVA on your dissimilarity matrix will provide you with the relevant statistics for assessing whether there is significant differences between your groups – keep in mind that the ordination plot is simply a way of visualising differences between samples.
To do this, we will use the adonis2 function from the vegan package, after first generating our dissimilarity matrix using the package's vegdist.
- Here we will show an example with Bray-Curtis distance, but if you were using 16S data and wanted to calculate the distance matrix with UniFrac distances, you could replace the
vegdistfunction with theUniFracfunction from thephyloseqpackage. - As far as the
adonis2function goes, the formula left-hand side takes a dissimilarity/distance matrix, and the right side takes the unquoted name(s) of columns from a data.frame (in this case the sample data object of our phyloseq object). As with other formulas in R, the right side can take multiple arguments, and can utilise both+and*notation.
# Generate the Bray-Curtis dissimilarity matrix (or the appropriate distance metric)
bact_bray_dist <- phyloseq::distance(bact_data_logCSS, method = 'bray')
# Run PERMANOVA
bact_bray_permanova <- adonis2(bact_bray_dist ~ group, data = data.frame(sample_data(bact_data_logCSS)), permutations = 1e4)This will provide an output that looks something like this:
| Df | SumOfSqs | R2 | F | Pr(>F) | |
|---|---|---|---|---|---|
| timeframe | 2 | 0.6863103 | 0.07398396 | 3.635218 | 0.001 |
| Residual | 91 | 8.5901631 | 0.92601604 | NA | NA |
| Total | 93 | 9.2764734 | 1.00000000 | NA | NA |
To investigate sample taxonomic composition, we can either look at the average composition of groups (using some sample metadata variable) or at the composition of each individual sample.
In either case, we will first need to filter the taxa using the microbiome::core() function to increase the clarity of the resulting barplots. We will place that within the phyloseq::tax_glom() function – to agglomerate to the genus level for example – and then transform the data to compositional form, and use the phyloseq::psmelt() function to pivot the data longer for plotting with ggplot2.
# Filter taxa to increase clarity of barplots
bact_data_tax <- psmelt(microbiome::transform(tax_glom(core(bact_data_filtered, detection = 10, prevalence = 0.2),
'Genus', NArm = T), transform = 'compositional', target = 'OTU'))Next, we will prepare a colour vector for the barplots using the RColorBrewer package.
# Prepare colours for plotting
library(RColorBrewer)
qual_col_pals <- brewer.pal.info[brewer.pal.info$category == 'qual',]
col_vector <- unlist(mapply(brewer.pal, qual_col_pals$maxcolors, rownames(qual_col_pals)))We will first plot the "average" barplot using a grouping variable. This could be a treatment group, time frame, etc. In this example, we will continue on with the drug treatment example from above, using the treatment as our grouping variable and also facet by time frame (correcting the facet labels by passing the facet_col_labels vector we created above to the labeller argument of facet_grid()).
# Plot taxonomic composition by group over time
(bact_taxonomy_gen <- ggplot(bact_data_tax, aes(x = group, y = Abundance, fill = Genus, NArm = FALSE)) +
geom_bar(stat = 'identity', position = 'fill', size = 0.01) +
scale_fill_manual(values = col_vector) + # use the colour vector we created
labs(title = 'Genera Distribution vs Treatment Group',
x = 'Treatment Group',
y = 'Abundance') +
theme(legend.key.size = unit(3.5, 'mm'), # change the size of each legend box
legend.position = 'bottom',
plot.margin = margin(0.25, 3, 0.25, 1.5, unit = 'cm'), # increase margins around the plot
axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1)) + # rotate x-axis labels
facet_grid(cols = vars(days_postTx), labeller = as_labeller(facet_col_labels))
)
ggsave(here::here('figures', 'composition', 'bact_taxonomy_genera_by_time.pdf'), bact_taxonomy_gen,
width = 20, height = 15, units = 'cm')The resulting output file looks like this:
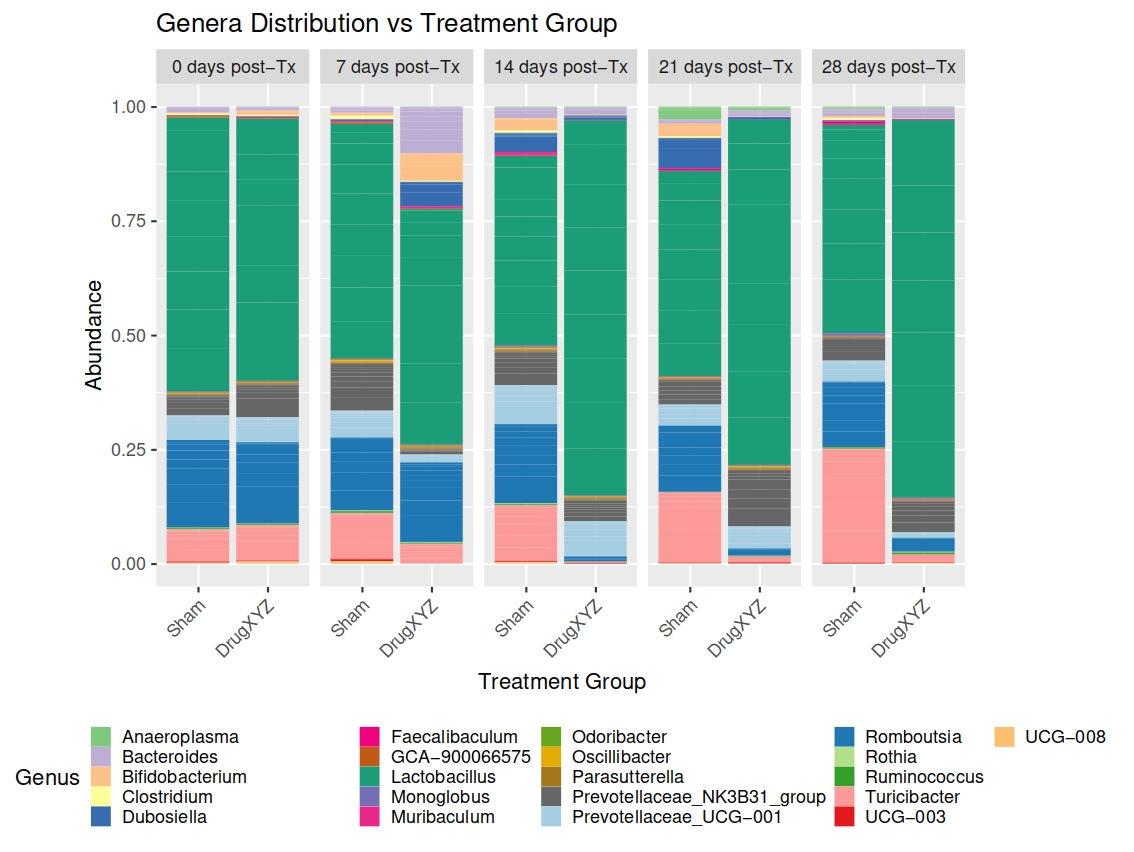
Seeing as we also know the IDs of the animals, we can also plot the taxonomic composition for each individual over time, facetting by both group and time frame.
We also need to add the treatment names to the end of the facet_col_labels vector here, because we are now facetting by two factors. The labeller just takes a single named vector for all labels.
# Barplot of abundance for each individual by group over time
(bact_taxonomy_gen_rat <- ggplot(bact_data_tax, aes(x = rat_id, y = Abundance, fill = Genus, NArm = FALSE)) +
geom_bar(stat = 'identity', position = 'fill', size = 0.01) +
scale_fill_manual(values = col_vector) +
labs(title = 'Genera Distribution vs Treatment Group',
x = 'Rat',
y = 'Abundance') +
theme(legend.key.size = unit(3.5, 'mm'),
legend.position = 'bottom',
plot.margin = margin(0.25, 3, 0.25, 2.25, unit = 'cm'),
axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1)) +
facet_grid(row = vars(days_postTx), cols = vars(group), scales = 'free', labeller = as_labeller(c(facet_col_labels,
'Sham' = 'Sham',
'DrugXYZ' = 'DrugXYZ')))
)
ggsave(here('figures', 'composition', 'bact_taxonomy_gen_rat_by_time.pdf'), bact_taxonomy_gen_rat,
width = 20, height = 20, units = 'cm')The resulting output file looks like this:

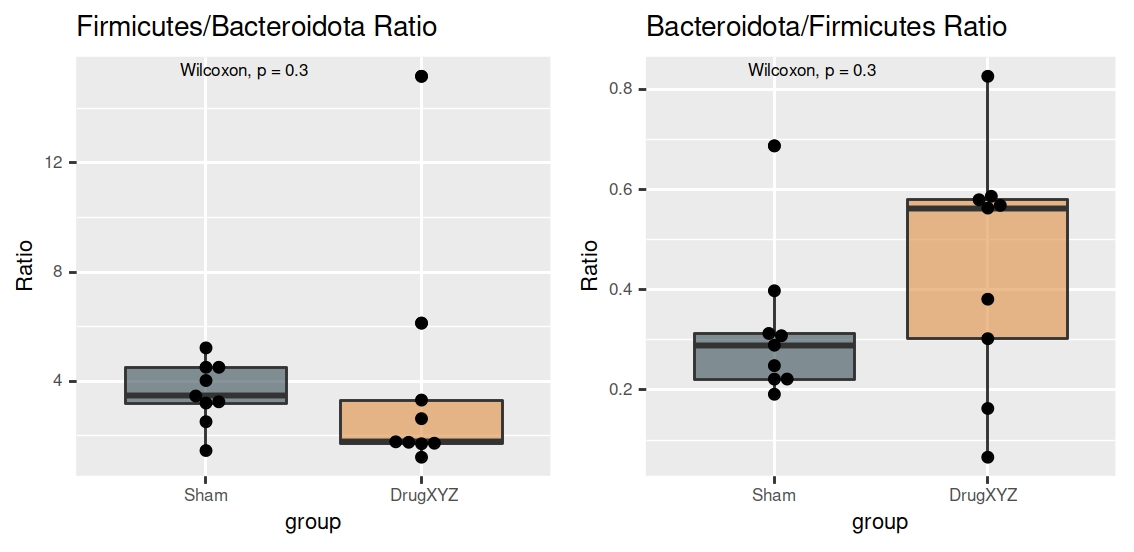
In particular the ratio between Firmicutes and Bacteroidetes is a popular measure, often used in assessing the health of the gastrointestinal microbiome. That said, we can look at the ratios of all phyla using the phyla_ratios() function we provide here.
-
phyloseq_object: aphyloseqobject to use for differential abundance testing. This should be aphyloseqobject containing raw (not normalised) counts. -
test_variable: the name of a single column from thesample_datato be used for comparison of phyla ratios. -
average_reads_threshold(default = 1000): optional - the minimum number of reads per sample on average that a phylum should have in order to be retained for analysis of ratios. Assessed by multiplying this value by the number of samples – if the row sums of a given phylum have less thanaverage_reads_threshold * number of samples, it will be removed. -
plot_output_folder: optional - the path to a folder where you would like output plots to be saved. If left blank, no plots will be saved. -
plot_file_prefix: optional - a string to attach to the start of the individual file names for your plots. This input is only used if theplot_output_folderargument is also provided.
-
phyla_ratios: a data.frame of the input data for each phylum-phylum comparison that passed thresholding, along with the ratio and test_variable. -
phyla_ratio_plots: aggplot2plot of each phylum-phylum comparison that passed thresholding, with either a Wilcoxon or Kruskal-Wallis statistic depending on the number of groups. -
stats_all: a data.frame containing all pair-wise Wilcoxon Rank Sum testing statistics for each comparison. -
stats_signif: a filtered version ofstats_allcontaining only those comparisons with a p-value < 0.5.
An example of the output .pdf file is shown here:
If you used this repository in a publication, please mention its url.
- Copyright (c) 2023 Mucosal Immunology Lab, Monash University, Melbourne, Australia.
- Authors: M. Macowan