LIONESS, or Linear Interpolation to Obtain Network Estimates for Single Samples, can be used to reconstruct single-sample networks (http://arxiv.org/pdf/1505.06440.pdf). This package implements the LIONESS equation in R to reconstruct single-sample networks. The default network reconstruction method we use here is based on Pearson correlation. However, lionessR can run on any network reconstruction algorithm that returns a complete, weighted adjacency matrix. To run the function on other network reconstruction algorithms, the user should substitute the function netFun with their network reconstruction algorithm of choice. lionessR works for both unipartite and bipartite networks and returns an R data frame that includes weights for all edges in each of the sample-specific networks.
The easiest way to install the R package lionessR is via the devtools package from CRAN:
install.packages("devtools")
library(devtools)
devtools::install_github("mararie/lionessR")
And then load the package using: library(lionessR).
Below we describe an example of co-expression network analysis using lionessR. This code is also available in the lionessR vignette.
As an example of how to model single-sample networks using lionessR, we will reconstruct and analyze co-expression networks for individual osteosarcoma patients. Osteosarcoma is an aggressive primary bone tumor that has a peak incidence in adolescents and young adults. The leading cause of death of osteosarcoma is distant metastases, which develop in 45% of patients. Detection of differential co-expression between osteosarcoma biopsies of patients with short and long metastasis-free survival (MFS) may help better understand metastasis development, and may point to targets for treatment.
Our example dataset contains gene expression data for pre-treatment biopsies of two groups of patients—one group contains 19 patients who did not develop any metastases within 5 years after diagnosis of the primary tumor (long-term metastasis-free survivors), the other group contains 34 patients who did developed metastases within 5 years (short-term metastasis-free survivors).
For this analysis, we need to load the following packages:
library(devtools)
library(lionessR)
library(igraph)
library(reshape2)
library(limma)
To start, we load the dataset in R:
data(OSdata)
This dataset contains an object exp with expression data for these 53 patients and an object targets with information on which patients developed metastases within 5 years. Because co-expression networks are usually very large, we subset this dataset to the 500 most variably expressed genes. To do this, we will sort the dataset based on the standard deviation of expression levels of each gene, and will select the top 500 genes:
nsel=500
cvar <- apply(as.array(as.matrix(exp)), 1, sd)
dat <- cbind(cvar, exp)
dat <- dat[order(dat[,1], decreasing=T),]
dat <- dat[1:nsel, -1]
dat <- as.matrix(dat)
Next, we will make two condition-specific networks, one for the short-term and one for the long-term MFS group. We calculate the difference between these condition-specific network adjacency matrices, so that we can use this to select edges that have large absolute differences in their co-expression levels:
groupyes <- which(targets$mets=="yes")
groupno <- which(targets$mets=="no")
netyes <- cor(t(dat[,groupyes]))
netno <- cor(t(dat[,groupno]))
netdiff <- netyes-netno
We use R packages igraph and reshape2 to convert these adjacency matrices to edgelists. As this is a symmetric adjacency matrix, we takeconvert the upper triangle of the co-expression adjacency matrix into an edge list. We then select those edges that have a difference in Pearson R correlation coefficient of at least 0.5:
cormat2 <- rep(1:nsel, each=nsel)
cormat1 <- rep(1:nsel,nsel)
el <- cbind(cormat1, cormat2, c(netdiff))
melted <- melt(upper.tri(netdiff))
melted <- melted[which(melted$value),]
values <- netdiff[which(upper.tri(netdiff))]
melted <- cbind(melted[,1:2], values)
genes <- row.names(netdiff)
melted[,1] <- genes[melted[,1]]
melted[,2] <- genes[melted[,2]]
row.names(melted) <- paste(melted[,1], melted[,2], sep="_")
tosub <- melted
tosel <- row.names(tosub[which(abs(tosub[,3])>0.5),])
Next, we’ll model the single-sample networks based on co-expression using lionessR. Note that, depending on the size of the dataset, this could take some time to run. We subset these networks to the selection of edges which we had defined above:
cormat <- lioness(dat, netFun)
row.names(cormat) <- paste(cormat[,1], cormat[,2], sep="_")
corsub <- cormat[which(row.names(cormat) %in% tosel),3:ncol(cormat)]
corsub <- as.matrix(corsub)
We then run a LIMMA analysis on these edges:
group <- factor(targets[,2])
design <- model.matrix(~0+group)
cont.matrix <- makeContrasts(yesvsno = (groupyes - groupno), levels = design)
fit <- lmFit(corsub, design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2e <- eBayes(fit2)
toptable <- topTable(fit2e, number=nrow(corsub), adjust="fdr")
We select the top 50 most differentially co-expressed edges and convert them into an igraph graph.data.frame object for visualization. We color edges red if they have higher coefficients in the short-term MFS group, and blue if they have higher coefficients in the long-term MFS group:
toptable_edges <- t(matrix(unlist(c(strsplit(row.names(toptable), "_"))),2))
z <- cbind(toptable_edges[1:50,], toptable$logFC[1:50])
g <- graph.data.frame(z, directed=F)
E(g)$weight <- as.numeric(z[,3])
E(g)$color[E(g)$weight<0] <- "blue"
E(g)$color[E(g)$weight>0] <- "red"
E(g)$weight <- 1
Next, we perform a LIMMA analysis on gene expression so that we can also color nodes based on their differential expression:
topgeneslist <- unique(c(toptable_edges[1:50,]))
fit <- lmFit(exp, design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2e <- eBayes(fit2)
topDE <- topTable(fit2e, number=nrow(exp), adjust="fdr")
topDE <- topDE[which(row.names(topDE) %in% topgeneslist),]
topgenesDE <- cbind(row.names(topDE), topDE$t)
We color nodes based on the t-statistic from the LIMMA analysis:
# add t-statistic to network nodes
nodeorder <- cbind(V(g)$name, 1:length(V(g)))
nodes <- merge(nodeorder, topgenesDE, by.x=1, by.y=1)
nodes <- nodes[order(as.numeric(as.character(nodes[,2]))),]
nodes[,3] <- as.numeric(as.character(nodes[,3]))
nodes <- nodes[,-2]
V(g)$weight <- nodes[,2]
# make a color palette
mypalette4 <- colorRampPalette(c("blue","white","white","red"), space="Lab")(256)
breaks2a <- seq(min(V(g)$weight), 0, length.out=128)
breaks2b <- seq(0.00001, max(V(g)$weight)+0.1,length.out=128)
breaks4 <- c(breaks2a,breaks2b)
# select bins for colors
bincol <- rep(NA, length(V(g)))
for(i in 1:length(V(g))){
bincol[i] <- min(which(breaks4>V(g)$weight[i]))
}
bincol <- mypalette4[bincol]
# add colors to nodes
V(g)$color <- bincol
Finally, we visualize these results in a network diagram of the 50 most significant edges from the LIMMA analysis:
par(mar=c(0,0,0,0))
plot(g, vertex.label.cex=0.7, vertex.size=10, vertex.label.color = "black", vertex.label.font=3, edge.width=10*(abs(as.numeric(z[,3]))-0.7), vertex.color=V(g)$color)
This will return the following image:
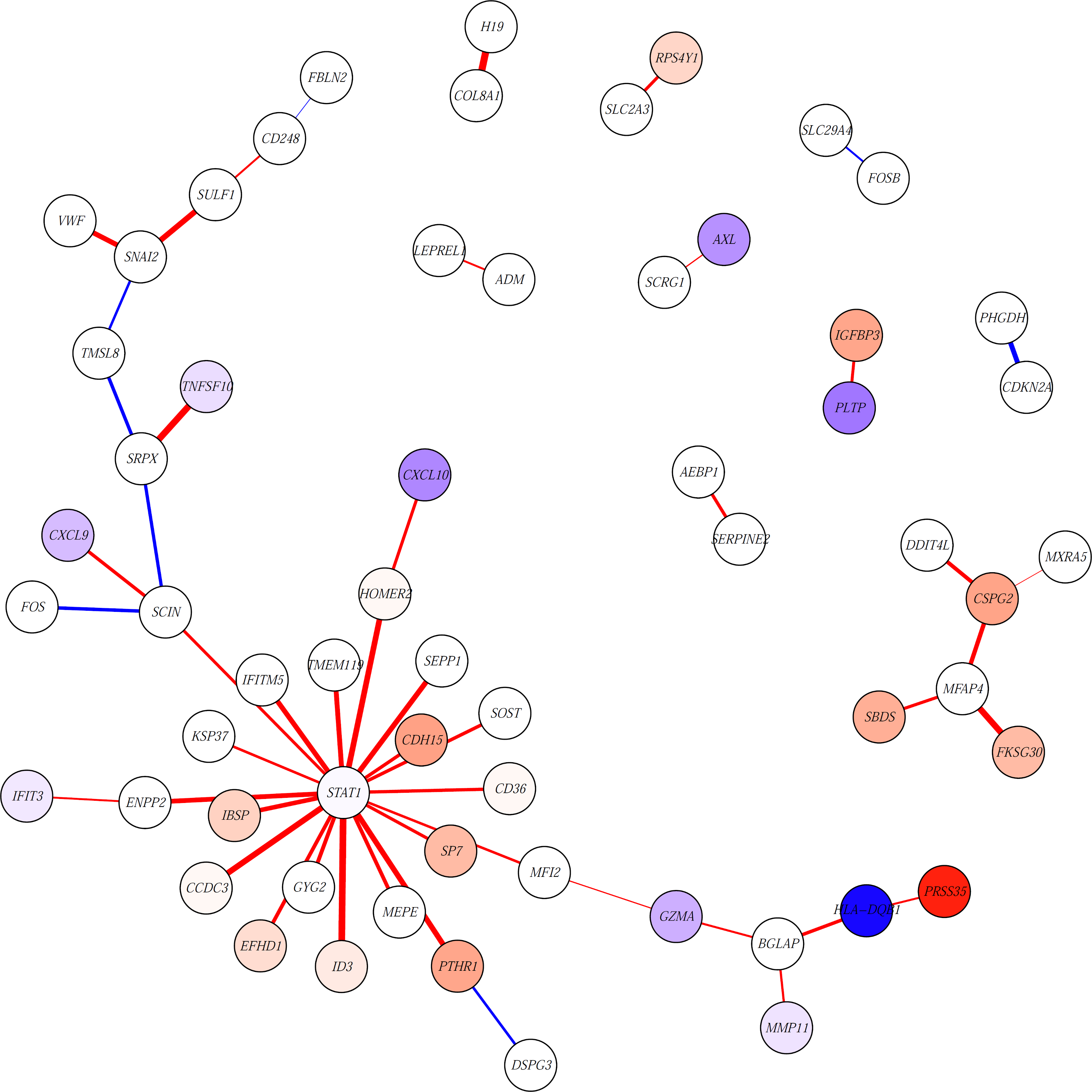
Here, edges are colored based on whether they have higher weights in patients with poor (red) or better (blue) MFS. Thicker edges represent higher log fold changes. Nodes (genes) are colored based on the t-statistic from the differential expression analysis. Nodes with absolute t-statistic < 1.5 are shown in white, nodes in red/blue have higher expression in patients with poor/better MFS, respectively.