The general idea of this tutorial is to get you familar with general bioinformatics. Much of the examples are pertinent to the Texas A&M University (TAMU) High Performance Research Compututing (HPRC) resources.
There are a few easy ways to access the TAMU HPRC.
Important:You must be physically on campus or logged into TAMU VPN to access the clusters
- Access using SSH. Open terminal on linux or macOS, cmd or powershell on Windows.
# terminal command
ssh [email protected]
ssh [email protected]
ssh [email protected]
-
You can use MobaXterm on windows to access the terminal easier just reduces having to type in password and it has a file manager like sidebar that allows for FTP.
-
You can also use the HPRC Open OnDemand portal https://portal.hprc.tamu.edu/
Side note: I like to use a combination of all three depending on what you are doing and ease of procedure.
Let's login to GRACE because I do most of my work on GRACE.
Moving around in linux
pwd # this tells you your current directory
cd # change directory
ls # list what is in current directory add -l to see list view
mv # move
mkdir # make directory
rm # remove BE CAREFUL with this one
cd .. # go one folder up
ls .. # look at contents one folder structure up
In order to load a program we need for analysis or any step we should run the following lines.
module spider R
ml spider R
module spider R/4.2.0
First we need to download all the files. Now the tool to do this is called fastq-dump which is part of the sra-tools from ncbi. What this does is downloads the accession numbers that you tell it to from NCBI. This code below will download SRR7060177 then compress the fasta file to .gz format.
fastq-dump SRR7060177 --gzip
Now you dont want to have to type all this in so we can utilize many cores and many instance of the HPRC to do this relatively quickly. However we cannot do this from the SSH login window or you will get a nasty email and you could be put in HPRC timeout for a week or two. So we need to create what is called a slurm job. All a slurm Job is file that asks the job scheduler for certain resources you want (i.e. cores, ram, runtime, etc.). Otherwise the rest of the slurm file is just a bash script. The specs you can ask for are located here https://hprc.tamu.edu/wiki/Grace:Batch
Below we are going to download the dataset from the paper https://acsess.onlinelibrary.wiley.com/doi/full/10.3835/plantgenome2018.02.0010
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=download_NCBI_sequences
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=48
#SBATCH --mem=350G
#SBATCH --time=02:00:00
#SBATCH --mail-type=ALL
ml GCC/10.2.0 OpenMPI/4.0.5 SRA-Toolkit/2.10.9 parallel/20210322 WebProxy
#mkdir $SCRATCH/tomatoes
#mkdir $SCRATCH/tomatoes/rawdata
cd $SCRATCH/tomatoes/rawdata
parallel -j 45 fastq-dump {} --gzip ::: SRR7060177 SRR7060178 SRR7060179 SRR7060180 SRR7060181 SRR7060182 SRR7060183 SRR7060184 SRR7060185 SRR7060186 SRR7060187 SRR7060188 SRR7060189 SRR7060190 SRR7060191 SRR7060192 SRR7060193 SRR7060194 SRR7060195 SRR7060196 SRR7060197 SRR7060198 SRR7060199 SRR7060200 SRR7060201 SRR7060202 SRR7060203 SRR7060204 SRR7060205 SRR7060206 SRR7060207 SRR7060208 SRR7060209 SRR7060210 SRR7060211 SRR7060212 SRR7060213 SRR7060214 SRR7060215 SRR7060216 SRR7060217 SRR7060218 SRR7060219 SRR7060220 SRR7060221 SRR7060222 SRR7060223 SRR7060224 SRR7060225 SRR7060226 SRR7060227 SRR7060228 SRR7060229 SRR7060230 SRR7060231 SRR7060232 SRR7060233 SRR7060234 SRR7060235 SRR7060236 SRR7060237 SRR7060238 SRR7060239 SRR7060240 SRR7060241 SRR7060242 SRR7060243 SRR7060244 SRR7060245 SRR7060246 SRR7060247 SRR7060248 SRR7060249 SRR7060250 SRR7060251 SRR7060252 SRR7060253 SRR7060254 SRR7060255 SRR7060256 SRR7060257 SRR7060258 SRR7060259 SRR7060260 SRR7060261 SRR7060262 SRR7060263 SRR7060264 SRR7060265 SRR7060266 SRR7060267 SRR7060268 SRR7060269 SRR7060270 SRR7060271 SRR7060272 SRR7060273 SRR7060274 SRR7060275 SRR7060276 SRR7060277 SRR7060278 SRR7060279 SRR7060280 SRR7060281 SRR7060282 SRR7060283 SRR7060284 SRR7060285 SRR7060286 SRR7060287 SRR7060288 SRR7060289 SRR7060290 SRR7060291 SRR7060292 SRR7060293 SRR7060294 SRR7060295 SRR7060296 SRR7060297 SRR7060298 SRR7060299 SRR7060300 SRR7060301 SRR7060302 SRR7060303 SRR7060304 SRR7060305 SRR7060306 SRR7060307 SRR7060308 SRR7060309 SRR7060310 SRR7060311 SRR7060312 SRR7060313 SRR7060314 SRR7060315 SRR7060316 SRR7060317 SRR7060318 SRR7060319 SRR7060320 SRR7060321 SRR7060322 SRR7060323 SRR7060324 SRR7060325 SRR7060326
Copy this text into a file and name it download.SLURM then execute by going to the folder where the slurm file is and sbatch download.SLURM
THIS FAILED because the compute nodes are not connected to the internet. IN the module load section we will add WebProxy and it should work. Also comment out the two mkdir lines since the folders already exist.
Next we will use two software, fastcq and multqc to look at the quality of all the samples we just downloaded.
If we are just doing one sample
fastqc SRR7060177.fasta.gz
This will produce an HTML file <--- click here to see
Then we will combine the all the fastqc reports using multiqc. Run the following line in the folder of the folder containing all the fastqc reports
multiqc .
This will produce an HTML file <--- click here to see
Let's try to run these steps in one sbatch job file
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=fastq_QC
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=48
#SBATCH --mem=350G
#SBATCH --time=02:00:00
#SBATCH --mail-type=ALL
mkdir $SCRATCH/tomatoes/fastqc
mkdir $SCRATCH/tomatoes/multiqc
ml load GCC/11.2.0 OpenMPI/4.1.1 FastQC/0.11.9-Java-11 MultiQC/1.12 parallel/20210722
cd $SCRATCH/tomatoes/rawdata
parallel -j 45 fastqc {} -o $SCRATCH/tomatoes/fastqc ::: *fastq.gz
cd $SCRATCH/tomatoes/multiqc
multiqc --interactive $SCRATCH/tomatoes/fastqc
What we see in the multiqc run is that the second read is pretty trash. These were paired end reads and since the quality of R2 is pretty bad we will just cut these out so we dont have bad data going forward. There is some weird thing going on with the quality of the cut site but for now we won't worry about that.
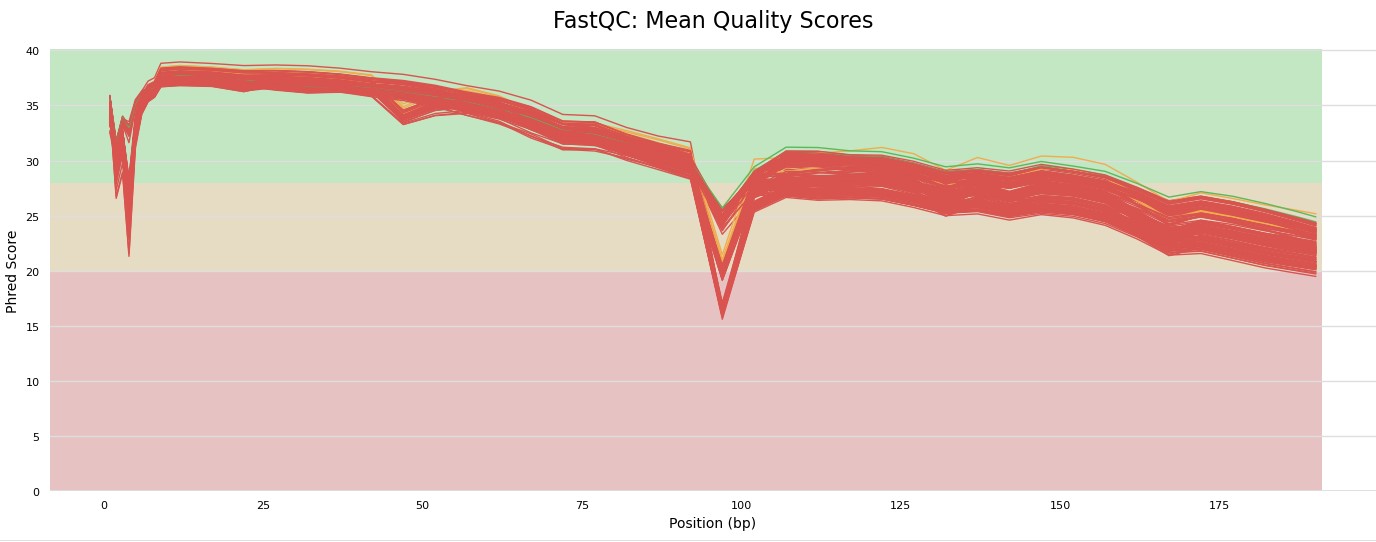
Because the quality drops off around 90 bp we will cut the reads here. we will use cutadapt. This program is also used to cut off adapters.
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=cutadapt
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=48
#SBATCH --mem=350G
#SBATCH --time=02:00:00
#SBATCH --mail-type=ALL
ml load GCC/11.2.0 parallel/20210722 cutadapt/3.5
mkdir $SCRATCH/tomatoes/trimmed
cd $SCRATCH/tomatoes/rawdata
parallel -j 10 cutadapt --cores=4 -l 90 -o $SCRATCH/tomatoes/trimmed/{} {} ::: *fastq.gz
After doing cutadapt this is homework assignment... write a SLURM job to do fastqc and multiqc on the new sequences that had the bad quality data trimmed off.
The next step is aligning the reads that we have cut to the reference genome. The general steps are:
- Pick a reference genome and download it. https://solgenomics.net/ftp/tomato_genome/assembly/
- I would just pick the most recent one. The paper uses an older reference
- https://solgenomics.net/ftp/tomato_genome/assembly/build_4.00/ choose the file that ends with .fa
- We will then need to create reference files for the alignment programs need for alignment.
# create a folder in the tomatoes folder called genome
mdir $SCRATCH/tomatoes/genome
cd $SCRATCH/tomatoes/genome
# download the reference genome
wget https://solgenomics.net/ftp/tomato_genome/assembly/build_4.00/S_lycopersicum_chromosomes.4.00.fa
or
curl https://solgenomics.net/ftp/tomato_genome/assembly/build_4.00/S_lycopersicum_chromosomes.4.00.fa -o S_lycopersicum_chromosomes.4.00.faThen we need to index the files. we will learn how to do this not submitting a job but for quick interactive sessions you can do this. To exit the srun ctrl+d
# open an interactive sbatch called srun this allows you do do computing from the terminal by passthrough to your terminal. Good for fast jobs that you want to make sure worked.
srun --nodes=1 --ntasks-per-node=4 --mem=100G --time=01:00:00 --pty bash -i
ml bwa-mem2/2.2.1-Linux64 picard/2.25.1-Java-11 GCC/12.2.0 SAMtools/1.17 WebProxy
cd $SCRATCH/tomatoes/genome
# indexing genome
bwa-mem2 index S_lycopersicum_chromosomes.4.00.fa
java -jar $EBROOTPICARD/picard.jar CreateSequenceDictionary -R S_lycopersicum_chromosomes.4.00.fa
samtools faidx S_lycopersicum_chromosomes.4.00.fabwa or bowtie typically used for alignments. we will use bwa-mem2 which is a faster multicore version of bwa
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=cutadapt
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=10
#SBATCH --mem=50G
#SBATCH --time=02:00:00
#SBATCH --mail-type=ALL
ml bwa-mem2/2.2.1-Linux64 GCC/12.2.0 SAMtools/1.17
cd $SCRATCH/tomatoes/trimmed
#mkdir /done
for file in *fastq.gz;
do
bwa-mem2 mem -t 8 $SCRATCH/tomatoes/genome/S_lycopersicum_chromosomes.4.00.fa $file | samtools view -b > $SCRATCH/tomatoes/bam/$file.bam
mv $file $SCRATCH/tomatoes/trimmed/done
done
After the reads are aligned, we need to sort the reads in the bam files. This sbatch below is for grace hprc cluster. For other clusters use ml spider parallel and ml spider picard to figure out what modules to load.
First cd to the directory where all the bam files are. Then we create a bash function which has the AddOrReplaceReadGroups and SortSam and remove intermediate file. then run this function in parallel
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=sortsam
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=64
#SBATCH --mem=250G
#SBATCH --time=24:00:00
#SBATCH --mail-type=ALL
ml GCCcore/11.2.0 parallel/20210722 picard/2.25.1-Java-11
cd /scratch/user/jzl0026/SWxBExMG/bam
sortsam_function(){
java -jar $EBROOTPICARD/picard.jar AddOrReplaceReadGroups -I $1 -O /scratch/user/jzl0026/fse/sorted_bam/$1.groups.bam -RGID $1 -RGLB NlaIII -RGPL ILLUMINA -RGPU unit1 -RGSM $1
java -jar $EBROOTPICARD/picard.jar SortSam I=/scratch/user/jzl0026/fse/sorted_bam/$1.groups.bam O=/scratch/user/jzl0026/fse/sorted_bam/$1.sorted.bam TMP_DIR=./tmp SORT_ORDER=coordinate CREATE_INDEX=true
rm /scratch/user/jzl0026/fse/sorted_bam/$1.groups.bam
}
export -f sortsam_function
parallel -j 60 sortsam_function {} {.} ::: *gz.bam
Now we are ready to run STACKS
We need to first make a file that has all the individuals in the file so that the program knows what files to expect and what population each individual belongs to. For our purpose, we will just count everything as one population.
In R, navigate to the folder where your sorted.bam files are. and setwd() there.
setwd("wherever your sorted bam files are")
sorted_bam = list.files(pattern="sorted.bam")
files_and_pop = data.frame(files=sorted_bam,pop="pop1")
files_and_pop$files = gsub(".sorted.bam",".sorted",files_and_pop$files )
write.table(files_and_pop,row.names = F,col.names = F, quote = F, file = "pop_map.tsv")Now ready to submit a job that runs stacks. There are two commands to execute(gstacks and populations). -I argument is the the folder where all the .sorted.bam files are, -M is the pop_map.tsv file we created in R. and -O is the output folder (create one that is called gstacks or something to hold the intermediate files between the two commands)
#!/bin/bash
#SBATCH --export=NONE
#SBATCH --job-name=gatk_parents
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=32
#SBATCH --mem=100G
#SBATCH --time=24:00:00
#SBATCH --mail-type=ALL
ml GCC/11.3.0 OpenMPI/4.1.4 Stacks/2.62
gstacks -I /scratch/user/jzl0026/SWxBExMG/sorted_bam -M /scratch/user/jzl0026/SWxBExMG/sorted_bam/pop_map.tsv -O /scratch/user/jzl0026/SWxBExMG/gstacks -t 32
populations -P /scratch/user/jzl0026/SWxBExMG/gstacks -O /scratch/user/jzl0026/SWxBExMG/stacks_population -t 32 --vcf
echo "done gstacks and populations"