Autoencoders are deep-learning based networks for dimension reduction and embedding by a combination of compressing encoder and decoder structure for non-linear and multi-modal data integration with promising application to complex biological data from large-scale omics measurements. Current ongoing research and publication provide many exciting architectures and implementations of autoencoders. However, there is a lack of easy-to-use and unified implementation covering the whole pipeline of autoencoder application.
Consequently, we present AUTOENCODIX with the following features:
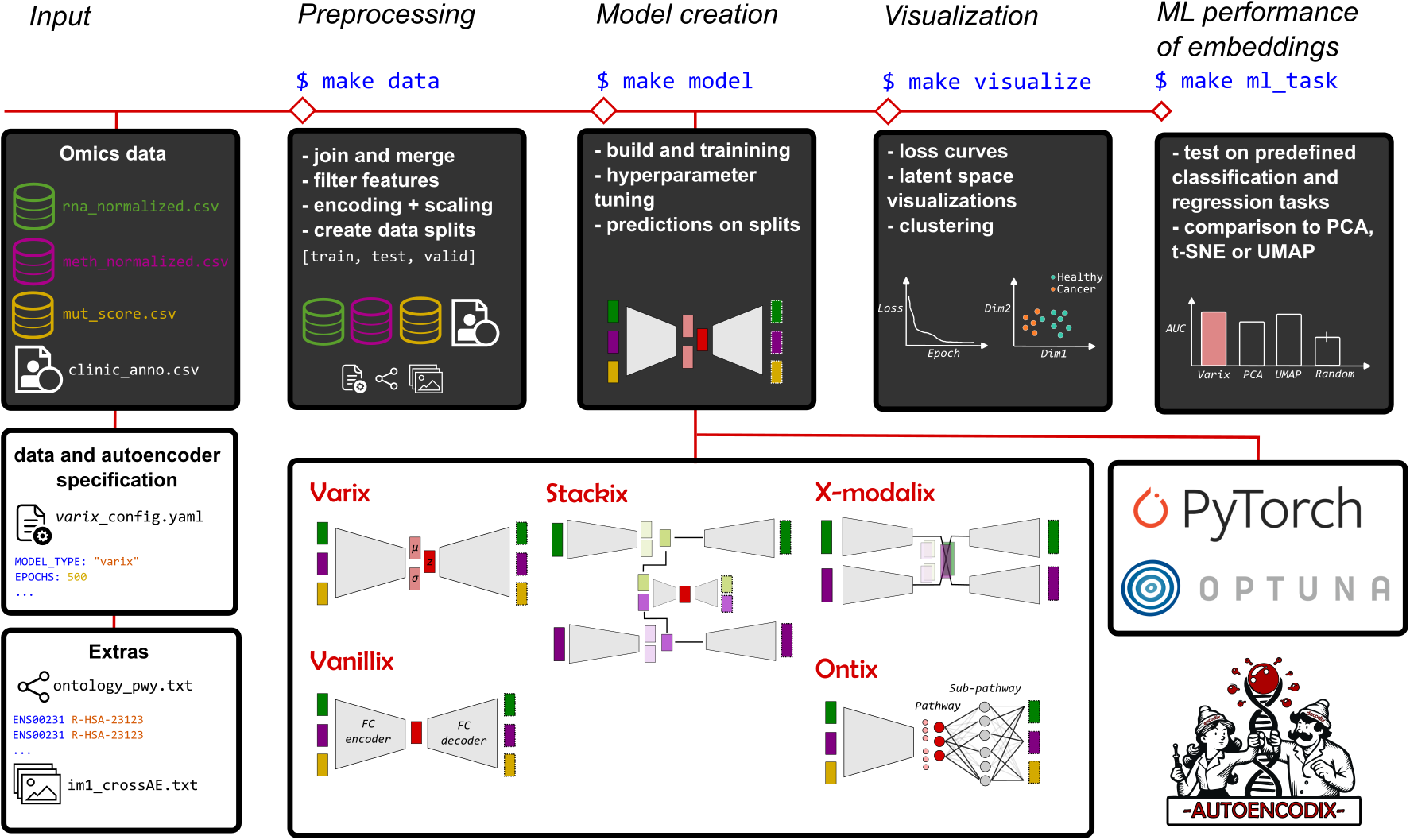
- Multi-modal data integration for any numerical or categorical data
- Different autoencoder architectures:
- vanilla
vanillix - variational
varix - hierarchical/stacked
stackix - ontology-based
ontix - cross-modal autoencoder (translation between different data modalities)
x-modalix
- vanilla
- A customizable set-up, run with your own data and change model parameters in a
yamlconfiguration file - Full pipeline from preprocessing to embedding evaluation:
For a detailed description and benchmark of capabilities, check our preprint and publication here: bioRxiv 10.1101/2024.12.17.628906
Please, use this to cite our work when using our framework:
@article {autoencodix2024,
author = {Joas, Maximilian and Jurenaite, Neringa and Pra{\v s}{\v c}evi{\'c}, Du{\v s}an and Scherf, Nico and Ewald, Jan},
title = {A generalized and versatile framework to train and evaluate autoencoders for biological representation learning and beyond: AUTOENCODIX},
year = {2024},
doi = {10.1101/2024.12.17.628906},
journal = {bioRxiv}
}
- AUTOENCODIX
- Table of contents
Follow the instructions depending on the machine you are working with. For familiarisation with the code, use your local machine on a small dataset as shown in our tutorials.
Requirements:
- pip
- Python == 3.10 (support for Python >=3.10 starting soon)
- GPU recommended for larger datasets
-
clone this repo
-
create environment with:
make create_environment -
activate env with
source venv-gallia/bin/activate -
install requirements with
make requirements
-
to use the Makefile in Windows you need to install
make -
Move
Makefile_windowstoMakefile -
create environment with:
make create_environment -
activate env with
.\venv-gallia\Scripts\activate -
install requirements with
make requirements -
if you encounter problems, see the troubleshooting section at the end
-
clone this repo
-
Overwrite
MakefilewithMakefile_macos -
activate env with
source venv-gallia/bin/activate -
install requirements with
make requirements -
currently GPU support is not available for MacOS
-
clone this repo in a dedicated space you want to decide to work in
-
load Python/3.10 or above and virtualenv
-
create a Python virtual environment according to your HPC guidelines
-
activate the environment with
source [env-name]/bin/activate -
install requirements with
make requirements
To work with our framework, only three steps are necessary:
- Get the input data ready
- Specify model and pipeline parameter via a
<run_id>_config.yaml-config file - Run the full pipeline via
make RUN_ID=<run_id>
First time users should check our tutorial notebooks for more details on those steps and showcases of important options of AUTOENCODIX:
- Framework set-up and input data preparation
- Basic config specification and VAE training
- Working with single-cell data and AUTOENCODIX
- Example of an ontology-based VAE providing explainability
- Showcase of cross-modal VAE for translation of gene expression to images
Additional to tutorial notebooks, we provide example configs of main features of AUTOENCODIX:
- Multi-modal VAE training on TCGA pan-cancer data
run_TCGAexample.shincluding hyperparameter tuning with Optuna via:
> ./bash-runs/run_TCGAexample.sh
- Training of an ontology-based VAE
ontixon single-cell data via:
> ./bash-runs/run_SingleCellExample.sh
All scripts will download the data, create necessary yaml-configs and run the pipeline for you. Results and visualizations can be found under reports/<run_id>/.
To work with our framework you first need to make sure that it has following format as described in details in the tutorial:
- for each data modality either a text-file (
csv,tsv,txt) or.parquet-file with samples as rows and features as columns - we recommend an
ANNOTATION-file in the same format containing clinical parameter or other sample meta-data for visualization - As described in the tutorials provide ontologies or image-mapping files to work with
ontixorx-modalix
When your input data is ready, you need to create your own config with the name <run_id>_config.yaml.
We provide a sample config in ./_config.yaml. Copy and rename this file:
> cp ./_config.yaml ./<RUN_ID>_config.yaml
Each entry in the _config.yaml has an inline comment that indicates whether you:
- Have to change this parameter (flagged with
TODO) - Should think about this parameter (flagged with
SHOULDDO) - probably don't need to change this parameter (flagged with
OPTIONAL)
All config parameters are explained in the _config.yaml file directly and the full documentation, here.
There are multiple steps in the pipeline. The steps can be run all at once, or step by step. To run the whole pipeline it is sufficient to run make ml_task RUN_ID=<run-id>.
If you only want to run all steps up until model training, you can run make model RUN_ID=<run-id>. This will prepare the data structure, process the data, and train your models. It is also possible to run steps without running the steps before. This can be done by the _only suffix. i.e. make model_only RUN_ID=<run-id>.
graph LR;
A[make ml_task]-->|also executes|B[make visualize];
B-->|also executes|C[make prediction];
C-->|also executes|D[make model];
D-->|also executes|E[make data];
E-->|also executes|F[make config];
After you have run the pipeline including make data and you want to make changes to model training parameters, you can re-run the pipeline with make train_n_visualize RUN_ID=<run-id>. This will skip data preprocessing and is useful is some cases when adjusting training parameters.
|-- bash-runs
| |-- run_slurm.sh
| |-- run_SingleCellExample.sh
| |-- run_TCGAexample.sh
|-- data
| |-- interim
| |-- processed
| | |-- <RUN_ID>
| | | |-- <data1.txt>
| | | |-- <data2.txt>
| | | `-- sample_split.txt
| |-- raw
| | |-- <raw_data.txt>
| | |-- images
| | | |---image_mappings.txt
| | | |---image1.jpg
|-- models
| |-- <RUN_ID>
| `-- <model.pt>
|-- reports
| |-- <RUN_ID>
| | |--<latent_space.txt>
| | |--figures
| | `--<figure1.png>
|-- src
| |-- data
| | |-- format_sc_h5ad.py
| | |-- format_tcga.py
| | |-- join_h5ad.py
| | |-- join_tcga.py
| | |-- make_dataset.py
| | `-- make_ontology.py
| |-- features
| | |-- build_features.py
| | |-- combine_MUT_CNA.py
| | `-- get_PIscores.py
| |-- models
| | |-- tuning
| | | |-- models_for_tuning.py
| | | |-- tuning.py
| | |-- build_models.py
| | |-- main_translate.py
| | |-- models.py
| | |-- predict.py
| | `-- train.py
| |-- utils
| | |-- config.py
| | |-- utils.py
| | `-- utils_basic.py
| |-- visualization
| | |-- Exp2_visualization.py
| | |-- Exp3_visualization.py
| | |-- ml_task.py
| | |-- vis_crossmodalix.py
| | `-- visualize.py
| |-- 000_internal_config.yaml
|-- ConfigParams.md
|-- LICENSE
|-- Makefile
|-- Makefile_macos
|-- README.md
|-- TCGAexample_config.yaml
|-- TuningExample_config.yaml
|-- _config.yaml
|-- clean.sh
|-- requirements.txt
|-- scExample_config.yaml
|-- setup.py
|-- test_environment.py
Running our pipeline will create as output multiple visualizations, latent space and reconstruction data frames and save PyTorch-models. Output can be found here:
reports/<run_id>/with visualizations and data frames of latent space (sample embeddings), loss metrics over epochs and other reportsmodels/<run_id>/PyTorch-model of final model as well as checkpoint-models if specified in the config.data/processed/<run_id>/all preprocessed input data and sample split file are stored.
The whole pipeline will automatically run on GPUs if GPUs are available (via torch.cuda.is_available() and using device cuda:0).
See our run_slurm.sh script for a sample slurm configuration with GPU. To run on an HPC cluster, you need to adjust the run_slurm.sh script with your paths. and then start the script like:
> bash ./bash-runs/run_slurm.sh <you-run-id>
This will send the sbatch script to your cluster. This way you can also easily parallelize multiple runs, by generating multiple config files.
We provide a clean script, clean.sh. The script either archives or deletes all folders of the given run id(s):
- data/processed
- data/interim
- models
- reports Run by:
> ./clean.sh -r <ID1,ID2,ID3,...,IDn>
will archive all output and interim data under archive as <run_id>.zip-files. If you want to delete, use the option -d. If you want to keep only the report folder and delete or archive the other, use the option -k.
For a more detailed benchmark of AUTOENCODIX check our preprint and publication under: https://www.biorxiv.org/
The implementation of autoencoder architectures is based and inspired by work of following authors and publications:
- For
vanillixandvarix: https://github.com/edianfranklin/autoencoder_for_cancer_subtype - For
varixandstackix: https://github.com/CancerAI-CL/IntegrativeVAEs - Ontology-based VAE
ontixwith sparse decoder: - cross-modal autoencoder
x-modalix:
Used data sets in notebooks and examples
- TCGA via cbioportal https://www.cbioportal.org/datasets
- pre-processed single-cell data via https://cellxgene.cziscience.com/
- in particular multiome RNA and ATAC-Seq data set of human cortex development https://doi.org/10.1126/sciadv.adg3754
- MNIST handwritten digits via KERAS https://keras.io/api/datasets/mnist/
- Images and proteom data of C. elegans embryogenesis https://doi.org/10.1038/s41592-021-01216-1
If you run for reproducibility with FIX_RANDOMNESS: "all" and you receive the following error:
RuntimeError: Deterministic behavior was enabled with either `torch.use_deterministic_algorithms(True)` or `at::Context::setDeterministicAlgorithms(true)`, but this operation is not deterministic because it uses CuBLAS and you have CUDA >= 10.2.
You need to run the following in your terminal before running our pipeline:
export CUBLAS_WORKSPACE_CONFIG=:16:8
If you run into the following error on Windows (Powershell):
error: Microsoft Visual C++ 14.0 or greater is required"
You can find solutions in this thread
Contributions are highly welcome, please check your guide