 |  |
Note (JFY): GeneSCF was recently (September 2020) moved to GitHub. It has been downloaded 5,325 times when hosted on http://genescf.kandurilab.org
Important: Always download latest stable release. To download latest release click here. Check here for other releases.
Gene Set Clustering based on Functional annotation. Most up-to-date and realtime information based gene enrichment analysis.
Publication:
GeneSCF: a real-time based functional enrichment tool with support for multiple organisms. BMC Bioinformatics 17, 365 (2016). https://doi.org/10.1186/s12859-016-1250-z
This documentation will provide detailed information on usage of GeneSCF tool (All versions). Read following page for running GeneSCF on test dataset.

- Overview
- Installation
- General usage
- Step-by-step instructions (simple usage)
- GeneSCF batch analysis
- License
GeneSCF is a command line based and powerful tool to perform gene enrichment analysis. GeneSCF uses realtime informatin from repositories such as geneontology, KEGG, Reactome and NCG while performing the analysis. This increases reliability of the outcome compared to other available tools. GeneSCF is command line tool designed to easily integrate with any next-generation sequencing analysis pipelines. One can use multiple gene list in parallel to save time. In simple terms,
- Real-time analysis, do not have to depend on enrichment tools to get updated.
- Easy for computational biologists to integrate this simple tool with their NGS pipeline.
- GeneSCF supports more organisms.
- Enrichment analysis for Multiple gene list in single run.
- Enrichment analysis for Multiple gene list using Multiple source database (GO,KEGG, REACTOME and NCG) in single run.
- Download complete GO terms/Pathways/Functions with associated genes as simple table format in a plain text file. Check Preparing database under Step-by-step instructions (simple usage) section for the details.
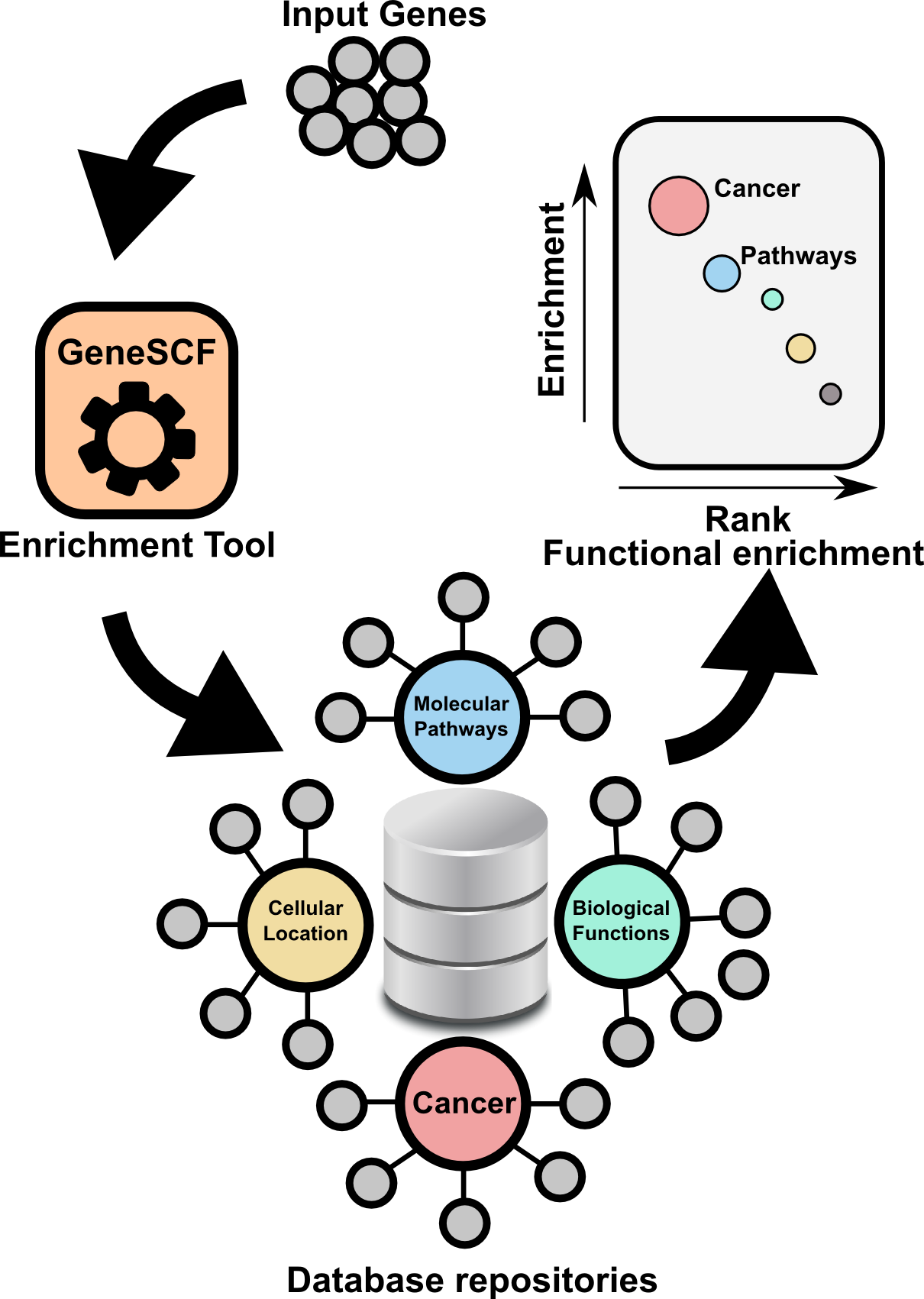
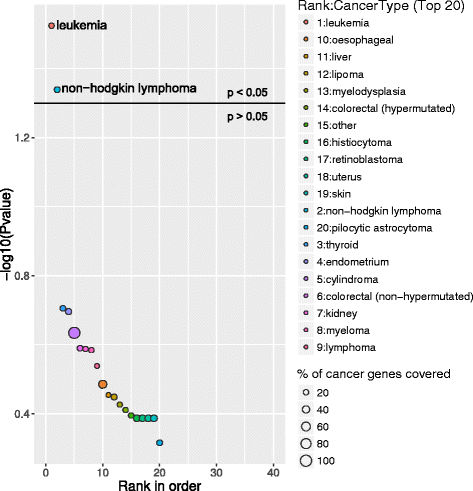
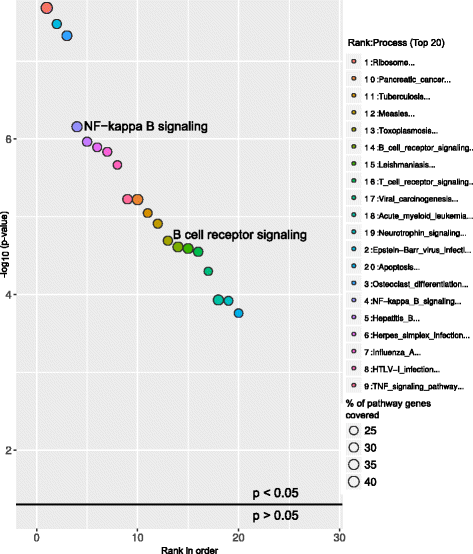
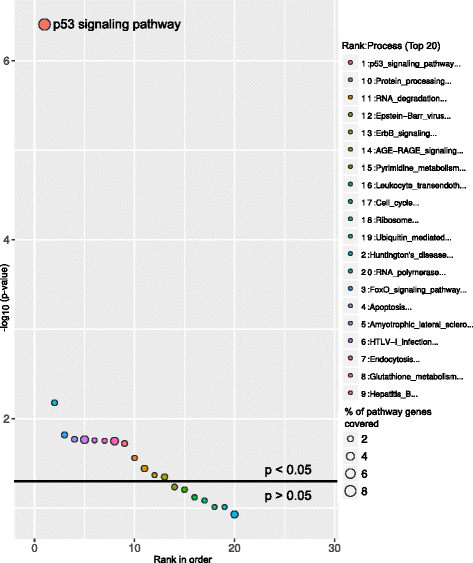
Above graphs are from Subhash and Kanduri et al., 2016
Download and extract the compressed file using 'unzip' (for .zip file) or 'tar' (for .tar.gz file). Use it without any need for special installation procedures.
- Dependencies: PERL
- Test required modules
: awk, cat, unzip, gzip, wget, rm, mkdir, sort, date, sed, paste, join, grep, curl, echo
- For graphical output or plots: If needed graphical output, pre installated R (version > 3.0) and 'ggplot2' is required.
Note: GeneSCF only works on Linux system, it has been successfully tested on Ubuntu, Mint and Cent OS. Other distributions of Linux might work as well.
There are two utilities available from GeneSCF package.
- One is the main command line 'geneSCF', to perform gene enrichment analysis.
- Next, is the 'prepare_database' command line to prepare the necessary database of an organism. GeneSCF by default comes with database for human consists of gene ontology, KEGG, Reactome, and NCG.
Note: Replace 'geneSCF-master-vx.x' from the commands with your GeneSCF directory.
./geneSCF-master-vx.x/geneSCF -m=[update|normal] -i=[INPUT FILE] -t=[gid|sym] -o=[OUTPUT PATH/FOLDER/] -db=[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME] -p=[yes|no] -bg=[#TotalGenes] -org=[see,org_codes_help]| Available Parameters in geneSCF | Options | Description |
|---|---|---|
| -m= | --mode= | normal update | For normal mode use normal and for update mode use *update* without quotes |
| -i= | --infile= | [INPUT-TEXT-FILE] | Input file contains list of Entrez GeneIDs or OFFICIAL GENE SYMBOLS.The genes must be new lines seperated (One gene per line) |
| -t= | --gtype= | gid sym | Type of input in the provided list either Entrez GeneIDs gid or OFFICIAL GENE SYMBOLS sym (default: *gid*) |
| -db= | --database= | GO_all GO_BP GO_CC GO_MF KEGG REACTOME NCG | Database to use as a source for finding gene enrichment, the options are either geneontology GO_all or geneontology-biological_process GO_BP or geneontology-molecular_function GO_MF or geneontology-cellular_components GO_CC or kegg KEGG or reactome REACTOME or Network of Cancer Genes NCG |
| -o= | --outpath= | [OUTPUT-DIRECTORY] | Existing directory to save output file (Don't forget to use trailing slash at end of the directory name). The output will be saved in the provided location as {INPUT_FILE_NAME}_{database}_functional_classification.tsv (tab-seperated file). **Note**: the specified folder should exist because GeneSCF does not create any output folder |
| -bg= | --background= | [Total background] | Total number of genes to be considered as background (Example : ~20,000 for human). It is important to choose the background appropriately. Sometimes your samples do not express all the genes. For example, if you are using differentially expressed genes for gene set enrichment analysis, you must choose total number of genes detected in your experiment including control and treatment samples irrespective of their differential status as your background (NOT the total number of genes in the annotation of the corresponding organism you are working with). All the genes from the annotation can be used when working with genes found in genome-wide studies (example, ChIP-seq, WGBS, etc.,). |
| -org= | --organism= | [see organism codes] | Please see organism codes (For human in KEGG ->hsa in Geneontology -> goa_human). For database 'REACTOME' and 'NCG', only human organism is supported in GeneSCF and the organism code is 'Hs'. |
| -p= | --plot= | yes no | For additional graphical output use yes or no.This requires R version > 3.0 and ggplot2 R package to be pre-installed on the system |
| -h | --help | For displaying this help page |
./geneSCF-master-vx.x/prepare_database -db=[GO_all|GO_BP|GO_MF|GO_CC|KEGG|REACTOME] -org=[see,org_codes_help directory]Note: The above command downloads complete '-db' of your choice as simple text file with corresponding genes per GO term in following location, 'geneSCF-master-vx.x/class/lib/db/[ORGANISM]/' for your prefered organism.
| Available Parameters in prepare_database | Options | Description |
|---|---|---|
| -db= | --database= | GO_all GO_BP GO_CC GO_MF KEGG REACTOME NCG | Database to use as a source for finding gene enrichment, the options are either geneontology GO_all or geneontology-biological_process GO_BP or geneontology-molecular_function GO_MF or geneontology-cellular_components GO_CC or kegg KEGG or reactome REACTOME |
| -org= | --organism= | [see organism codes] | Please see organism codes (For human in KEGG ->hsa in Geneontology -> goa_human). For database 'REACTOME' and 'NCG', only human organism is supported in GeneSCF and the organism code is 'Hs'. |
For a convenience we will use test datasets from the directory 'geneSCF-master-vx.x/test/'. There are two steps involved,
- Prepare your prefered database for an organism of your interest.
- Perform enrichment analysis on your gene list.
- One can also perform enrichment analysis in single-step using 'update' mode.
./geneSCF-master-vx.x/prepare_database -db=GO_all -org=goa_humanNote: Specific dabases can be also updated using 'GO_BP', 'GO_MF' and 'GO_CC'. The above command downloads complete geneontology ('GO_all') with corresponding genes per GO term as simple text file in following location, 'geneSCF-master-vx.x/class/lib/db/goa_human/'.
./geneSCF-master-vx.x/prepare_database -db=KEGG -org=hsa./geneSCF-master-vx.x/prepare_database -db=REACTOME -org=HsNote: Reactome supports only Human (Hs)
./geneSCF-master-vx.x/prepare_database -db=NCG -org=HsNote: NCG supports only Human (Hs)
./geneSCF-master-vx.x/geneSCF -m=normal -i=geneSCF-master-vx.x/test/H0.list -o=geneSCF-master-vx.x/test/output/ -t=sym -db=GO_BP -bg=20000 --plot=yes -org=goa_human./geneSCF-master-vx.x/geneSCF -m=normal -i=geneSCF-master-vx.x/test/H0.list -o=geneSCF-master-vx.x/test/output/ -t=sym -db=KEGG -bg=20000 --plot=yes -org=hsaNote: All predicted results can be found in 'geneSCF-master-vx.x/test/output/' folder with file name '{INPUT_FILE_NAME}_{database}_functional_classification.tsv'
This '-m=update' mode will integrate both 'prepare_database' and 'geneSCF' into single command mode. When you use 'update' mode once, you can use 'normal' mode for the next consecutive runs, in case you are planning to use the same database for different gene lists (This saves time).
./geneSCF-master-vx.x/geneSCF -m=update -i=geneSCF-master-vx.x/test/H0.list -o=geneSCF-master-vx.x/test/output/ -t=sym -db=GO_BP -bg=20000 --plot=yes -org=goa_human**Note:** The above command also downloads complete geneontology biological processes ('GO_BP') with corresponding genes per GO term as a simple text file in following location, '<b>geneSCF-master-vx.x/class/lib/db/goa_human/</b>' and also does enrichment analysis in parallel. The results for enrichment analysis can be found in folder '<b>geneSCF-master-vx.x/test/output/</b>'.
./geneSCF-master-vx.x/geneSCF -m=update -i=geneSCF-master-vx.x/test/H0.list -o=geneSCF-master-vx.x/test/output/ -t=sym -db=KEGG -bg=20000 --plot=yes -org=hsaNote: All predicted results can be found in 'geneSCF-master-vx.x/test/output/' folder with file name '{INPUT_FILE_NAME}_{database}_functional_classification.tsv'
Edit file './geneSCF-master-vx.x/db_batch_config.txt' to configure your parameters for batch run. The sample file looks like the one below,
#database:organism:background:type
#GO_BP:goa_human:20000:sym
#GO_MF:goa_human:20000:sym
GO_CC:goa_human:20000:sym
KEGG:hsa:20000:sym
#REACTOME:Hs:20000:sym
NCG:Hs:20000:symIn the above file you are asking GeneSCF to run enrichment analysis using 'GO_CC', 'KEGG', and 'NCG' database for human. The database mentioned with preceeding '#' will not be considered for the run.
Edit script './geneSCF-master-vx.x/geneSCF_batch' for your input files (files_path) and output path (output_path).
files_path="/FOLDER/WHERE/GENE_LISTS/STORED"
output_path="/FOLDER/PATH/FOR/OUTPUT"Note:
- It is recommended to keep all input files in same folder.
- Inside specified output folder path GeneSCF will automatically create individual sub-folders for each gene list.
Execute batch analysis.
./geneSCF-master-vx.x/geneSCF_batch