
![]()


- It modularizes the consensus clustering processes that various methods can be easily integrated in different steps of the analysis.
- It provides rich visualizations for intepreting the results.
- It allows running multiple methods at the same time and provides functionalities to compare results in a straightforward way.
- It provides a new method to extract features which are more efficient to separate subgroups.
- It generates detailed HTML reports for the complete analysis.
Zuguang Gu, et al., cola: an R/Bioconductor package for consensus partitioning through a general framework, Nucleic Acids Research, 2021. https://doi.org/10.1093/nar/gkaa1146
cola is available on Bioconductor, you can install it by:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cola")The latest version can be installed directly from GitHub:
library(devtools)
install_github("jokergoo/cola")Examples for cola analysis can be found at https://jokergoo.github.io/cola_examples/ and https://jokergoo.github.io/cola_collection/.
Online documentation is at https://jokergoo.github.io/cola.
Supplementary for the cola manuscript is at https://github.com/jokergoo/cola_supplementary and the scripts are at https://github.com/jokergoo/cola_manuscript.
There are the following vignettes:
- A Quick Start of Using cola Package
- A Framework for Consensus Partitioning
- Automatic Functional Enrichment on Signature genes
- Predict Classes for New Samples
- Work with Big Datasets
- Compare Partitions
- ATC - More Forms
- Hierarchical consensus partitioning
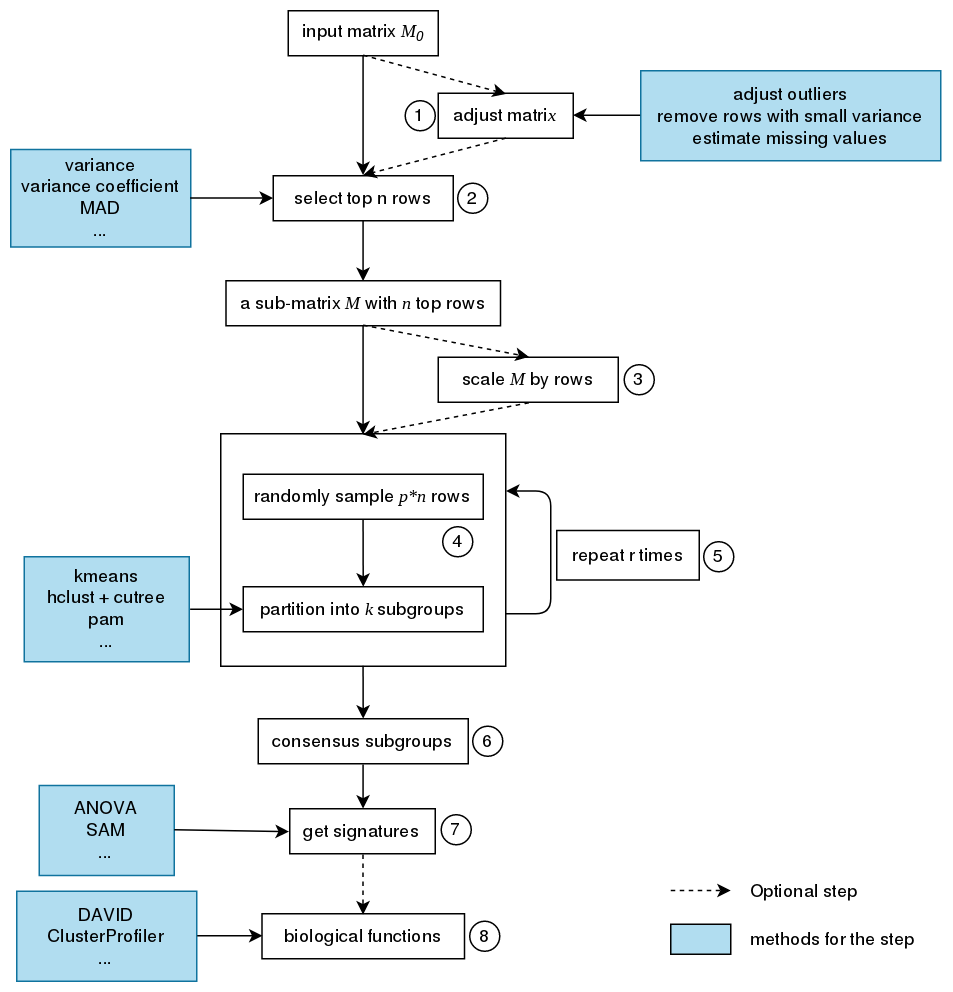
The steps of consensus partitioning is:
- Clean the input matrix. The processing are: adjusting outliers, imputing missing values and removing rows with very small variance. This step is optional.
- Extract subset of rows with highest scores. Here "scores" are calculated by
a certain method. For gene expression analysis or methylation data
analysis,
$n$ rows with highest variance are used in most cases, where the "method", or let's call it "the top-value method" is the variance (byvar()orsd()). Note the choice of "the top-value method" can be general. It can be e.g. MAD (median absolute deviation) or any user-defined method. - Scale the rows in the sub-matrix (e.g. gene expression) or not (e.g. methylation data). This step is optional.
- Randomly sample a subset of rows from the sub-matrix with probability
$p$ and perform partition on the columns of the matrix by a certain partition method, with trying different numbers of subgroups. - Repeat step 4 several times and collect all the partitions.
- Perform consensus partitioning analysis and determine the best number of subgroups which gives the most stable subgrouping.
- Apply statistical tests to find rows that show significant difference between the predicted subgroups. E.g. to extract subgroup specific genes.
- If rows in the matrix can be associated to genes, downstream analysis such as function enrichment analysis can be performed.
Three lines of code to perfrom cola analysis:
mat = adjust_matrix(mat) # optional
rl = run_all_consensus_partition_methods(
mat,
top_value_method = c("SD", "MAD", ...),
partition_method = c("hclust", "kmeans", ...),
cores = ...)
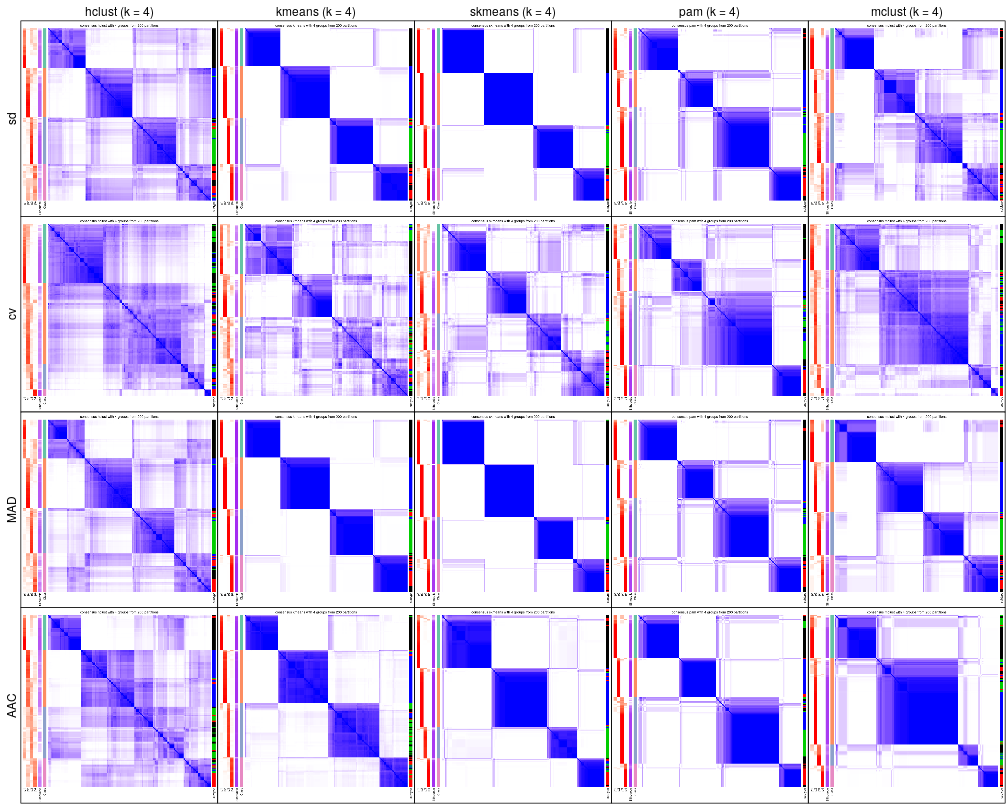
cola_report(rl, output_dir = ...)Following plots compare consensus heatmaps with k = 4 under all combinations of methods.
MIT @ Zuguang Gu
The cola icon in logo is made by photo3idea_studio from www.flaticon.com.