The Popcycle pipeline performs 3 different analyses:
- Filtration of Optimally Positioned Particles (OPP)
- Manual Gating of cytometric populations (VCT)
- Aggregate statistics for the different populations.
The metadata and aggregated statistics for each step are saved into a SQL database using SQLite3 and the particle data is saved into gzip compressed binary and text files.
First we need to satisfy some dependencies. Make sure Python 2.7 is installed with Anaconda. Then install the seaflowpy Python package:
git clone https://github.com/armbrustlab/seaflowpy
cd seaflowpy
python setup.py install
# To test your installation
python setup.py test
Now download and install the popcycle R library
git clone https://github.com/uwescience/popcycle
cd popcycle
# This will install popcycle and automatically run some tests
Rscript setup.R
Documentation for popcycle functions used in this guide can be accessed with the built-in R help system. e.g. running the following command in R will bring up a separate doc window for the filter.evt.files function.
?filter.evt.filesFirst start R and load the popcycle library
library(popcycle)Next, set a few variables which will define input and output file locations as well a cruise name. We'll call this cruise "testcruise" and use a small test SeaFlow data set that was installed with popcycle.
cruise <- "testcruise"
db <- paste0(cruise, ".db")
# For this guide we'll use an example data set that's installed
# with popcycle, but for real analysis this path would point to
# an EVT directory for a real cruise.
evt.dir <- system.file("extdata/SeaFlow/datafiles/evt", package="popcycle")
opp.dir <- paste0(cruise, "_opp")
vct.dir <- paste0(cruise, "_vct")db is the SQLite3 database file which will store aggregate statistics for OPP and VCT files as well as the filtering and gating parameters used to generate OPP and VCT data.
evt.dir is the input directory containing EVT files of raw, unfiltered particle data.
opp.dir is the output directory which will contain OPP files of filtered particle data.
vct.dir is the output directory which will contain VCT files of per particle population classifications
Create the popcycle SQLite3 database. If you're using a database that already exists - for example if you filtered using filterevt.py from the seaflowpy project - this step isn't necessary, but won't hurt anything either.
make.popcycle.db(db) # Create a popcycle SQLite3 database fileFor many database operations it's necessary to access the cruise information contained in SFL files, so let's load that now. We can load all SFL files found in the EVT directory or load from a single concatenated SFL file.
save.sfl(db, cruise, evt.dir=evt.dir)
# OR from a single file
# save.sfl(db, cruise, sfl.file=sfl.file)
get.sfl.table(db) # View SFL table to confirm importSeaFlow data is intially saved as EVT binary files representing three-minute windows of time. For example, in our example data set the EVT files within evt.dir are:
2014_185/2014-07-04T00-00-02+00-00.gz
2014_185/2014-07-04T00-03-02+00-00.gz
2014_185/2014-07-04T00-06-02+00-00.gz
2014_185/2014-07-04T00-09-02+00-00.gz
2014_185/2014-07-04T00-12-02+00-00.gz
This is actually a file path that includes the julian day directory, but it will be often be referred to as the file name in popcycle. A slightly modified version of this file name (no .gz extension) will be used to identify associated entries in the SQLite3 database and to name derived files for OPP and VCT data.
For example, the first file 2014_185/2014-07-04T00-00-02+00-00.gz would have a file field value of 2014_185/2014-07-04T00-00-02+00-00 in any database tables. The derived OPP and VCT files would be named 2014_185/2014-07-04T00-00-02+00-00.opp.gz and 2014_185/2014-07-04T00-00-02+00-00.vct.gz within the opp.dir and vct.dir output directories.
Any version of the file name can be converted to the short version used by popcycle functions with clean.file.path.
The fastest way to filter EVT files is to use seaflowpy_filter from the seaflowpy project. This will create filtered OPP file and database output equivalent to the R code in this section, but could potentially save you hours of time. It's possible to run this script on the command-line or through the seaflowpy_filter wrapper function in R. For example, to filter EVT files using 4 threads:
seaflowpy_filter(db, cruise, evt.dir, opp.dir, process.count=4, width=0.5, offset=0)Once this step is done it's possible to continue on to the Gating section. The rest of this section deals with filtering in R.
Set parameters for filtration and filter raw data to create OPP. In most cases it's sufficient to use default parameters.
save.filter.params(db) # Save default filter parameters
get.filter.params.latest(db) # Examine the default parameters we just setThis saves a log entry of new filter parameters. To set different parameters run save.filter.params again with custom parameters. Each time this function is run a new filter parameter entry is made in the database. By default the latest parameters are used for filtering, but it's possible to use a specific parameter set by filter ID.
To view all filter parameter entries and find filter IDs run
get.filter.table(db)Now we'll filter EVT files to create OPP data.
evt.files <- get.evt.files(evt.dir) # Find 5 EVT files in evt.dir
# Filter particles
filter.evt.files(db, cruise, evt.dir, evt.files, opp.dir)There should be three new OPP files of filtered particles in the testcruise_opp directory.
It is sometimes desirable to apply different filter parameters to different groups of EVT files. The filter parameters to use can be specified by the filter.id parameter to filter.evt.files.
# If you don't want to use the latest filter parameters, pass a
# filter ID retrieved from `get.filter.table(db)` to filter.evt.files
# e.g.
filter.evt.files(db, cruise, evt.dir, evt.files, opp.dir,
filter.id="d3afb1ea-ad20-46cf-866d-869300fe17f4")To get a subset of EVT files selected by date, use get.evt.files.by.date.
Now we're ready to set the gating for the different populations.
WARNINGS:
- The order in which you gate the different populations is very important, choose it wisely.
- The gating has to be performed over optimally positioned particles (opp), not over EVT files.
We want gating parameters that will be applicable to most opp files from the cruise. To optimize your chance of setting the proper gating parameters, do not set them based on one randomly chosen file. Instead, merge/combine as many files as you computer can handle. In this example, we are going to combine 20 files spaced evenly throughout the duration of the cruise.
opp.list <- get.opp.files(db) # get the list of all OPP files from the cruise
freq <- round(seq(75, length(opp.list), length.out=20))
OPP <- NULL
for (i in freq) {
opp.name <- opp.list[i]
opp <- get.opp.by.file(opp.dir, opp.name)
OPP <- rbind(OPP, opp)
}The first population to gate is always the beads, which are used as an internal standard for the instrument. Since the bead's optical properties remain relatively stable over time, we use a manual gating method to classify the population based on fsc_small and pe.
Note: This will open an R plot for a cytogram of the OPP file that consists of the appended data from the 20 opp files. Draw a polygon around the population (Left click to draw segment, right-click to close the segment and finalize the polygon).
gates.log <- add.manual.classification(OPP, "beads", "fsc_small", "pe")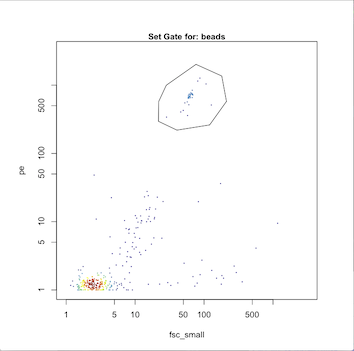
Once the polygon for the beads has been drawn, we can start setting the gates for phytoplankton population:
WARNING: The order in which you gate the different populations is very important
- Start by gating the
Synechococcuspopulation based onfsc_smallandpe - After that, gate the
Prochlorococcuspopulation basedfsc_smallandchl_small. - Finally, gate the
Picoeukaryotepopulation basedfsc_smallandchl_small.
Note: Since the optical properties of a particular phytoplankton population can change dramatically over time and space, so you may need to use different sets of polygons to properly gate your population of interest. If manual gating is too much trouble, there is a more automated gating approach:
In the case of Synechococcus and Prochlorococcuspopulation, we use a semi-supervised algorithm (modified from flowDensity package) to draw ellipses around the population using the function add.auto.classification().
This function breaks the cytogram plot into 4 quadrants by density. Using the parameter position=c(), select the quadrant where you observe the population. Use the examples params provided for gates, scale, and min.pe, but play around with them until you see optimal results. Make sure to append the updated classification onto the current by using the gates.log=gates.log.
gates.log <- add.auto.classification("synecho", "fsc_small", "pe",
position=c(FALSE,TRUE), gates=c(3.0,NA),
scale=0.975, min.pe=3, gates.log=gates.log)
gates.log <- add.auto.classification("prochloro", "fsc_small", "chl_small",
position=c(FALSE,TRUE), gates=c(2.0,0.5),
scale=0.95, gates.log=gates.log)Once the parameters are defined for Synechococcus and Prochlorococcuspopulation, use manual gating to cluster all the phytoplankton cells left (namely Picoeukaryote population) using manual.classification. Don’t worry about overlapping populations inside of your polygon. Since this is the last population we are gating, all other gated population will not be included.
gates.log <- add.manual.classification(OPP, "picoeuk", "fsc_small", "chl_small", gates.log)You can test your various gating parameters on the subset of files to ensure accuracy before saving the gating parameters. In this case, we will use a subset of 20 evenly distributed files and display the output of the classification on a cytogram.
opp.list <- get.opp.files(db) # get the list of all OPP files from the cruise
freq <- round(seq(75, length(opp.list), length.out=20))
par(mfrow=c(4,5), mar = c(1.75, 1.5, 1.5, 1.5), oma = c(0.5, 0.5, 0.5, 0.5))
for(i in freq){
opp.name <- opp.list[i]
opp <- get.opp.by.file(opp.dir, opp.name)
opp <- try(classify.opp(opp, gates.log))
try(plot.vct.cytogram(opp, para.x="fsc_small", para.y="chl_small", main=paste(basename(opp.name))))
}Once satisfied, save the gating in the db. Similar to the save.filter.params function, save.gating.params saves the gating parameters and order in which the gating was performed. Every call to save.gating.params creates a new gating entry in the database which can be retrieved by ID. Then, save the classification. (Note, when redo-doing classification, make sure to always set the gating params for “beads” first. If you skip this step, gates.log will not be reset, and you classification will still contain the parameters wished to be changed).
gating.id <- save.gating.params(db, gates.log)
classify.opp.files(db, cruise, opp.dir, opp.list, vct.dir, gating.id=gating.id)Manual classification performance can be increased by using the Python package seaflowpy. The script seaflowpy_classify can be run on the command-line or through the R wrapper function of the same name. e.g. to classify using 4 threads:
seaflowpy_classify(db, cruise, opp.dir, vct.dir, process.count=4,
gating.id="c3a06970-3552-4c1c-a71d-f71af93f4d60")To classify a subset of files the start.file and end.file parameters can be used to specify the subset of files to classify.
WARNING The auto-classification on Synechococcus and Prochlorococcuspopulation requires the flowDensity R package, and it will NOT work in python. So use seaflowpy_classify for Manual gating only.
Once clustering has finished you can obtain the aggregate statistics for the whole cruise with
stat <- get.stat.table(db)Data can be plotted using a set of functions:
-
To plot the filter steps
evt.files <- get.evt.files(evt.dir) evt.name <- evt.files[2] # to select the 2nd evt file of the list plot.filter.cytogram.by.file(evt.dir, evt.name)
-
To plot an evt cytogram. WARNING: the number of particles in an evt file can be high (>10,000) which can be a problem for some computers. We advise to limit the disply to < 10,000 particles.
evt.files <- get.evt.files(evt.dir) evt.name <- evt.files[2] # to select the 2nd evt file of the list plot.evt.cytogram.by.file(evt.dir, evt.name) # TO LIMIT the number of displyed particles to 10,000 evt <- readSeaflow(file.path(evt.dir, evt.name)) if(nrow(evt) > 10000) evt <- evt[round(seq(1, nrow(evt), length.out=10000)),] plot.cytogram(evt, "fsc_small","chl_small")
-
To plot an opp cytogram
# OPTION 1: SELECT OPP data by FILES opp.files <- get.opp.files(db) opp.name <- opp.files[2] # to select the 2nd opp file opp <- get.opp.by.file(opp.dir, opp.name) plot.cytogram(opp, "fsc_small","chl_small") # OR DIRECTLY plot.opp.cytogram.by.file(opp.dir, opp.name, "fsc_small","chl_small") # OPTION 2: SELECT OPP data by DATE # e.g. select 10 min of data opp <- get.opp.by.date(db, opp.dir, "2014-07-04 00:00", "2014-07-04 00:10") plot.cytogram(opp, "fsc_small","chl_small")
-
To plot an opp cytogram with clustered populations
opp.files <- get.opp.files(db) opp.name <- opp.files[2] # to select the 2nd opp file plot.vct.cytogram.by.file(opp.dir, vct.dir, opp.name)
-
To plot aggregate statistics, e.g., cell abundance the cyanobacteria
Synechococcuspopulation on a map or over time. Unfortunately, with our small example data set these figures are faily underwhelming.stat <- get.stat.table(db) # to load the aggregate statistics plot.map(stat, pop='synecho', param='abundance') plot.time(stat, pop='synecho', param='abundance')
But you can plot any parameter/population, just make sure their name matches the one in the 'stat' table...
FYI, type
colnames(stat)to know which parameters are available in thestattable, andunique(stat$pop)to know the name of the different populations. -
Data stored in the popcycle.db can be examined directly in R. To view any table, run the corresponding
get.<table>.table(db)function. For exampleget.sfl.table(db) get.filter.table(db) get.gating.table(db) get.poly.table(db) get.opp.table(db) get.vct.table(db) # stat is not actually a table, but rather the result of a joined # query between the sfl, opp, and vct tables. However, for our purposes # here it will be considered as a table. get.stat.table(db)