Nextflow pipeline to perform BAM realignment or fastq alignment, with/without local indel realignment and base quality score recalibration.
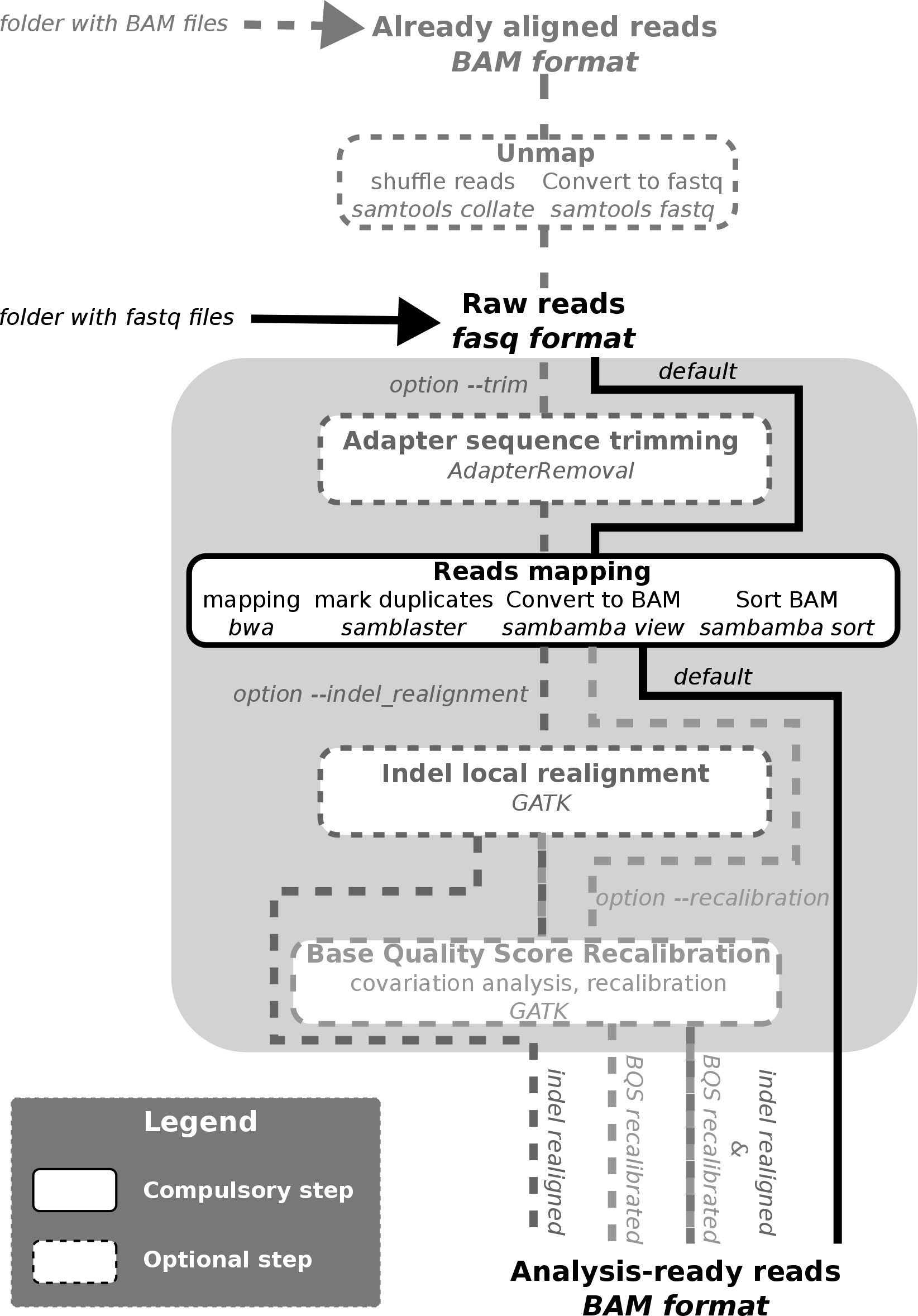
workflow
For the basic fastq files alignment without indel realignment, base
quality score recalibration, nor alternative contif handling, the script
requires the following programs: -
*nextflow*. Install with
bash curl -fsSL get.nextflow.io | bash And move it to a location
in your $PATH (e.g., /usr/local/bin):
bash sudo mv nextflow /usr/local/bin -
*bwa* -
*samblaster* -
*sambamba* and the
following files: - a fasta reference genome with its index files
(.fai, .sa, .bwt, .ann, .amb, .pac; in the same directory) -
a folder with fastq files
In addition, for the bam files realignment: - *samtools* - a folder with bam files
For the adapter sequence trimming: - *AdapterRemoval*
For the ALT contigs handling, additional softwares and scripts are required: - the k8 javascript execution shell (e.g., available in the *bwakit* archive) - javascript bwa-postalt.js and the additional fasta reference .alt file from *bwakit* must be in the same directory as the reference genome file.
For the indel realignment: - GATK *GenomeAnalysisTK.jar* - GATK bundle VCF files with lists of indels (recommended: 1000 genomes and Mills gold standard VCFs)
For the base quality score recalibration: - GATK *GenomeAnalysisTK.jar* - GATK bundle VCF files with lists of indels and SNVs (recommended: 1000 genomes indels, Mills gold standard indels VCFs, dbsnp VCF) - bed file with intervals to be considered
nextflow run iarcbioinfo/alignment-nf --input_folder input --fasta_ref hg19.fasta --out_folder outputTo use the adapter trimming step, you must add the *--trim* option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf --input_folder input --fasta_ref reference/hs38DH.fa -out_folder output --trimTo use the alternative contigs handling mode, you must provide the path to an ALT aware genome reference (e.g., hg38) AND add the *--alt* option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf --input_folder input --fasta_ref reference/hs38DH.fa --js /user/bin/k8/k8 --postaltjs /user/bin/bwa-0.7.15/bwakit/bwa-postalt.js -out_folder output --altTo use the local indel realignment step, you must provide the path to the GATK jar file, the GATK bundle folder, AND add the *--indel_realignment* option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf --GATK_bundle GATKbundle/hg19 --input_folder input --fasta_ref reference/hg19.fa --GATK_folder /user/bin7GATK-3.6-0 --out_folder output --indel_realignmentTo use the base quality score recalibration step, you must provide the path to the GATK jar file, the GATK bundle folder, a bed file, AND add the *--recalibration* option, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/alignment-nf --GATK_bundle GATKbundle/hg19 --input_folder input --fasta_ref reference/hg19.fa --GATK_folder /user/bin7GATK-3.6-0 --intervals reference/hg19_intervals.bed --out_folder output --recalibration| **PARAMETER* * | DEFAULT | **DESCRIPTION* * |
|---|---|---|
| --help | null | print usage and optional parameters |
| --input_fo lder | . | input folder |
| --fasta_re f | hg19.fasta | genome reference |
| --cpu | 8 | number of CPUs |
| --mem | 32 | memory |
| --mem_samb amba | 1 | memory for software sambamba |
| --RG | PL:ILLUMINA | sequencing information for aligned (for bwa) |
| --fastq_ex t | fastq.gz | extension of fastq files |
| --suffix1 | _1 | suffix for second element of read files pair |
| --suffix2 | _2 | suffix for second element of read files pair |
| --out_fold er | . | output folder for aligned BAMs |
| *--intervals * | bed file with interval list | |
| --GATK_bun dle | bundle | path to GATK bundle files |
| --GATK_fol der | . | path to GATK GenomeAnalysi sTK.jar file |
| --trim | false | enable adapter sequence trimming |
| --indel_re alignment | false | perform local indel realignment (GATK) |
| --recalibra tion | false | perform quality score recalibration (GATK) |
| --js | k8 | path to javascript interpreter k8 |
| *--postaltjs * | bwa-postalt.js" | path to postalignment javascript bwa-postalt.j s |
| --alt | false | enable alternative contig handling (for reference genome hg38) |