RNAseq mapping, quality control, and reads counting nextflow pipeline
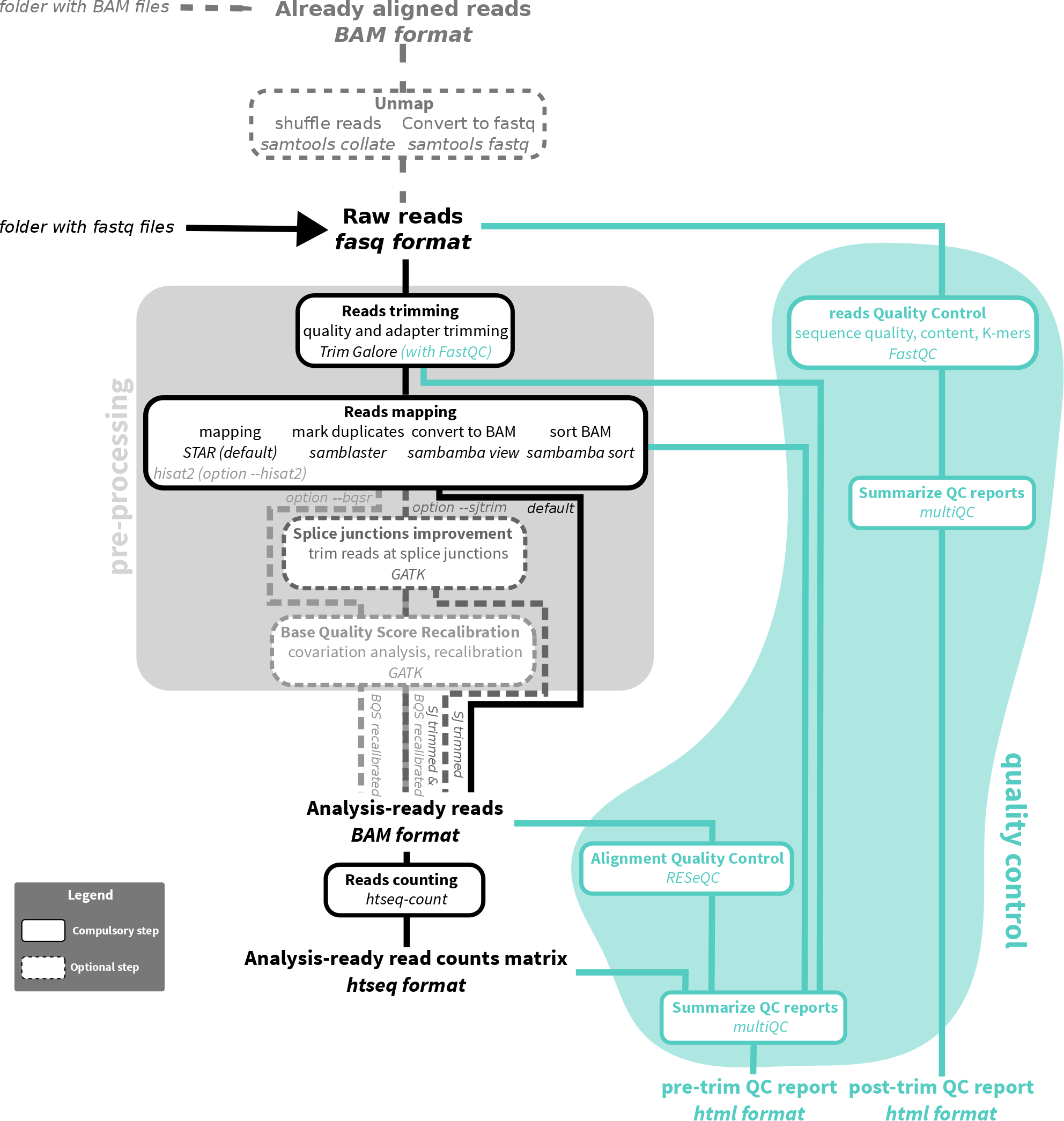
workflow
The following programs need to be installed and in the PATH environment variable: - *fastqc* - *cutadapt*, which requires Python version > 2.7 - *trim_galore* - *RESeQC* - *multiQC* - *STAR* - *htseq*; the python script htseq-count must also be in the PATH - *nextflow*
In addition, STAR requires genome indices that can be generated from a genome fasta file ref.fa and a splice junction annotation file ref.gtf using the following command:
STAR --runThreadN n --runMode genomeGenerate --genomeDir ref --genomeFastaFiles ref.fa --sjdbGTFfile ref.gtf --sjdbOverhang 99In order to perform the optional alignment with hisat2, hisat2 must be installed: - *hisat2*
In addition, indexes files .ht2 must be downloaded from generated from *hisat2*, or generated from a reference fasta file (e.g., reference.fa) and a GTF annotation file (e.g., reference.gtf) using the following commands:
extract_splice_sites.py reference.gtf > genome.ss
extract_exons.py reference.gtf > genome.exon
hisat2-build reference.fa --ss genome.ss --exon genome.exon genome_tranIn order to perform the optional reads trimming at splice junctions, GATK must be installed: - GATK *GenomeAnalysisTK.jar*
In addition, index .fai and dictionnary .dict must be generated from the fasta reference genome using the following commands:
samtools faidx ref.fa
java -jar picard.jar CreateSequenceDictionary R= ref.fa O= ref.dict- GATK *GenomeAnalysisTK.jar*
- GATK bundle VCF files with lists of indels and SNVs (recommended: 1000 genomes indels, Mills gold standard indels VCFs, dbsnp VCF)
- bed file with intervals to be considered
To run the pipeline on a series of paired-end fastq files (with suffixes *_1* and *_2*) in folder *fastq*, and a reference genome with indexes in folder *ref_genome*, one can type:
nextflow run iarcbioinfo/RNAseq-nf --input_folder fastq --gendir ref_genome --suffix1 _1 --suffix2 _2To use the reads trimming at splice junctions step, you must add the *--hisat2* option, specify the path to the folder containing the hisat2 index files, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/RNAseq-nf --input_folder fastq --suffix1 _1 --suffix2 _2 --hisat2 --hisat2_idx /home/user/reference/genome_tranTo use the reads trimming at splice junctions step, you must add the *--sjtrim* option, specify the path to the folder containing the GenomeAnalysisTK jar file, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/RNAseq-nf --input_folder fastq --gendir ref_genome --suffix1 _1 --suffix2 _2 --sjtrim --GATK_folder /home/user/GATKTo use the base quality score recalibration step, you must add the *--bqsr* option, specify the path to the folder containing the GenomeAnalysisTK jar file, the path to the GATK bundle folder for your reference genome, specify the path to the bed file with intervals to be considered, as well as satisfy the requirements above mentionned. For example:
nextflow run iarcbioinfo/RNAseq-nf --input_folder fastq --gendir ref_genome --suffix1 _1 --suffix2 _2 --bqsr --GATK_folder /home/user/GATK --GATK_bundle /home/user/GATKbundle --intervals intervals.bed| **PARAMETER* * | DEFAULT | **DESCRIPTION* * |
|---|---|---|
| --help | null | print usage and optional parameters |
| --input_fo lder | . | input folder |
| --output_f older | . | output folder |
| --gendir | ref | reference genome folder |
| --cpu | 4 | number of CPUs |
| --mem | 50 | memory for mapping |
| --memOther | 2 | memory for QC and counting |
| --fastq_ex t | fq.gz | extension of fastq files |
| --suffix1 | _1 | suffix for second element of read files pair |
| --suffix2 | _2 | suffix for second element of read files pair |
| --output_f older | . | output folder for aligned BAMs |
| --annot_gt f | Homo_sapiens.GR Ch38.79.gtf | annotation GTF file |
| --annot_gf f | Homo_sapiens.GR Ch38.79.gff | annotation GFF file |
| --fasta_re f | ref.fa | reference genome fasta file for GATK |
| --GATK_fol der | GATK | folder with jar file GenomeAnalysis TK.jar |
| --GATK_bun dle | GATK_bundle | folder with files for BQSR |
| *--intervals * | intervals.bed | bed file with intervals for BQSR |
| --RG | PL:ILLUMINA | string to be added to read group information in BAM file |
| --sjtrim | false | enable reads trimming at splice junctions |
| --bqsr | false | enable base quality score recalibration |
| *--gene_bed * | gene.bed | bed file with genes for RESeQC |
| --stranded | no | Strand information for counting with htseq [no, yes, reverse] |
| --stranded | no | Strand information for counting with htseq [no, yes, reverse] |
| --hisat2 | false | use hisat2 instead of STAR for mapping |
| --hisat2_i dx | genome_tran | index filename prefix for hisat2 |