Home
See Paper

iBrowser can be downloaded as an self contained (Virtual Box) virtual machine using BitTorrent Sync and this read-only key: key (860Mb). A manual can be found here
The data used in the iBrowser paper can be downloaded using BitTorrent Sync using the following read-only key: key.
The data consists of:
total 39G 9.1G arabidopsis_50k.sqlite 1.5K arabidopsis_50k.sqlite.nfo 6.4G RIL_10k.sqlite 1.5K RIL_10k.sqlite.nfo 1.5K RIL_50k_mode_ril_delete_greedy.sqlite.nfo 1.6G RIL_50k_mode_ril_delete.sqlite 1.5K RIL_50k_mode_ril_delete.sqlite.nfo 995M RIL_50k_mode_ril_greedy.sqlite 1.5K RIL_50k_mode_ril_greedy.sqlite.nfo 1.6G RIL_50k_mode_ril.sqlite 1.5K RIL_50k_mode_ril.sqlite.nfo 1.6G RIL_50k.sqlite 1.5K RIL_50k.sqlite.nfo 1.5K RIL.customorder 67M tom84_10k_introgression.sqlite 1.5K tom84_10k_introgression.sqlite.nfo 11G tom84_10k.sqlite 1.5K tom84_10k.sqlite.nfo 27M tom84_50k_introgression.sqlite 1.5K tom84_50k_introgression.sqlite.nfo 3.1G tom84_50k.sqlite 1.5K tom84_50k.sqlite.nfo 3.0K tom84.customorder 3.9G tom84_genes.sqlite 1.5K tom84_genes.sqlite.nfo
The Web User Interface (Web UI) works in Chrome 37+ and Firefox 33+. The UI behaviour in iExplorer is erratic and therefore we discourage the use of this web browser. We haven't tested Opera and Safari web browsers, feedback is welcome.
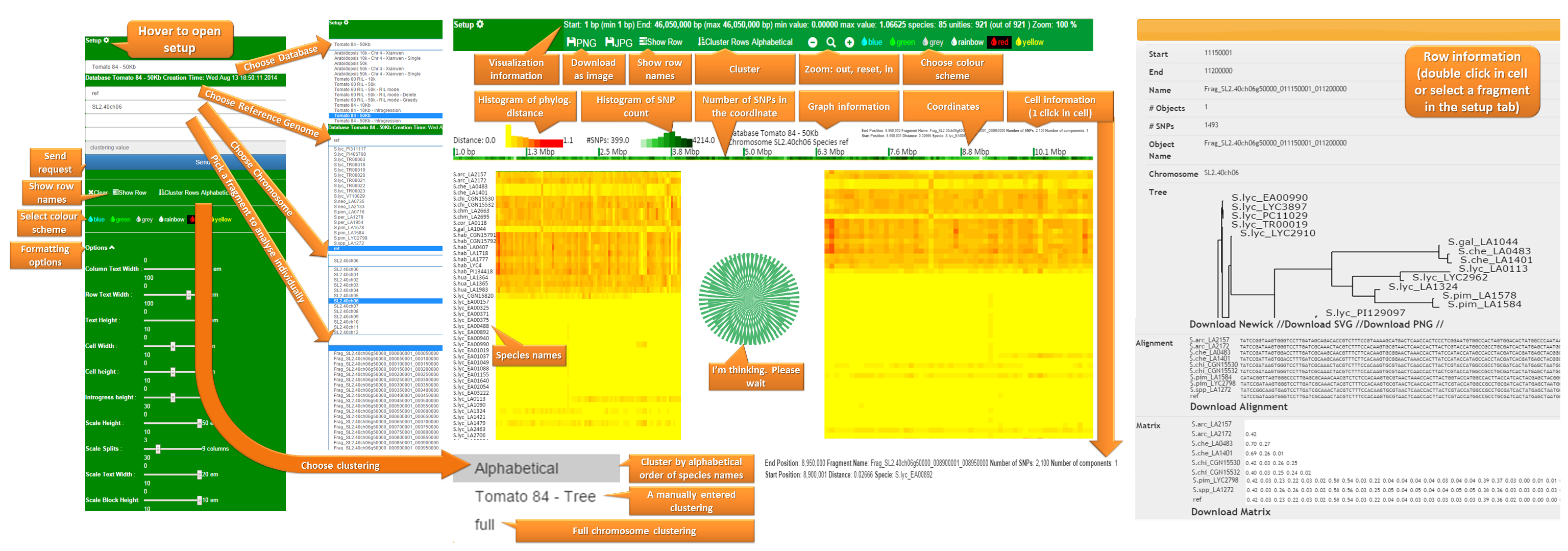
for standalone:
docker run -it --rm \ -v $PWD/data:/var/www/ibrowser/data \ -v $PWD/access.log:/var/log/apache2/access.log \ -v $PWD/error.log:/var/log/apache2/error.log \ -p 127.0.0.1:10000:10000 \ --name ibrowser \ sauloal/introgressionbrowser
for local copy,run:
git clone [email protected]:sauloal/introgressionbrowser.git cd introgressionbrowser docker run -it --rm \ -v $PWD:/var/www/ibrowser \ -v $PWD/data:/var/www/ibrowser/data \ -v $PWD/access.log:/var/log/apache2/access.log \ -v $PWD/error.log:/var/log/apache2/error.log \ -p 127.0.0.1:10000:10000 \ --name ibrowser \ sauloal/introgressionbrowser_local
Open your browser at 127.0.0.1:10000
replace 127.0.0.1 for 0.0.0.0 in the command line if you want others in your network to be able to access your iBrowser instance.
replace the -it for -d to run in the background
The virtual machine should run automatically. The only procedure is to share your data folder (in your host computer) as "DATA". A step-by-step manual can be found here. In case you want/need to do it manually, please find bellow instructions:
wget http://download.virtualbox.org/virtualbox/4.3.6/VBoxGuestAdditions_4.3.6.iso mkdir vbox mount VBoxGuestAdditions_4.3.6.iso vbox cd vbox ./VBoxLinuxAdditions.run cd .. umount vbox edit /etc/fstab adding data /media/data vboxsf re 0 0 mount -a ls /media/data
currently there's a bug in vmware which doesn't allows for the mounting of shared folders. For this reason vmware is not currently supported
mkdir /mnt/cdrom mount /dev/cdrom /mnt/cdrom mkdir ~/vm cd ~/vm tar xvf /dev/cdrom/VMwareTools-9.6.1-1378637.tar.gz cd vmware-tools-distrib ./vmware-install.pl -d cd ../.. rm -rf vm ls /mnt/hgfs/data
Clone or download Introgression Browser.
git clone https://github.com/sauloal/introgressionbrowser
Install Linux dependencies:
apt-get install -y -f libapache2-mod-wsgi apache2 nano build-essential \
checkinstall openssl sqlite3 libsqlite3-dev \
libfreetype6 libfreetype6-dev zlib1g-dev libjpeg62 libjpeg62-dev \
pkg-config libblas-dev liblapack-dev gfortran zlib1g-dev
apt-get install -y -f python-setuptools python-dev python-numpy python-scipy \
python-matplotlib python-pandas python-sympy python-pip python-imaging \
python-numpy pylint
Install python dependencies:
pip install --requirement requirements.txt
OR
easy_install --user flask easy_install --user ete2 easy_install --user sqlalchemy easy_install --user Flask-SQLAlchemy easy_install --user pysha3 easy_install --user pycrypto
If not possible to install python libraries system wide with apt-get, install also:
easy_install --user Pillow easy_install --user Image easy_install --user numpy easy_install --user scipy easy_install --user matplotlib easy_install --user MySQL-python
Install pypy (Optional, but speeds up analysis)
Copy config.template to your data directory:
cp config.template [PATH TO DATA FOLDER]/config.py
Edit config.py to configure:
- whether user control is enables or not (Default: False)
- the server's port (Default: 10000)
- pages which can be seen without login (to disable, comment line with #)
- enable/disable debug of server
#decide whether to have user control or not
HAS_LOGIN = False
#define port to server webpage
SERVER_PORT = 10000
# pages which can be seen without login
librepaths = [
'/api',
'/favicon.ico'
]
DEBUG = False
Initialize iBrowser by running:
./ibrowser.py [PATH TO DATA FOLDER] init
This will
- Create session secret
- Create RSA key
- create SSL certificate
- Create default user (admin:admin)
ONLY IN CASE YOU WANT ACCESS CONTROL
- Edit config.py and Set hasLogin to "True"
- (ADVISED) Change admin password (otherwise default admin:admin will be created) by running:
./ibrowser.py [PATH TO DATA FOLDER] deluser admin ./ibrowser.py [PATH TO DATA FOLDER] adduser admin [DESIRED PASSWORD]
-
Optional
-
(Default: 2048) Change RSA key size by editing [PATH TO DATA FOLDER]/config.keylen
echo 2048 > [PATH TO DATA FOLDER]/config.keylen
- Clean all config by running:
./ibrowser.py [PATH TO DATA FOLDER] clean
- Create users manually by running (can be perfomed in the UI):
./ibrowser.py [PATH TO DATA FOLDER] adduser [USER] [DESIRED PASSWORD]
- Delete users manually by running (can be perfomed in the UI):
./ibrowser.py [PATH TO DATA FOLDER] deluser [USER]
- List users by running (can be perfomed in the UI):
./ibrowser.py [PATH TO DATA FOLDER] listusers
Run ibrowser.py
./ibrowser.py [PATH TO DATA FOLDER]
This set of scripts takes as input a series of Variant Call Files (VCF) of species mapped against a single reference. After a series of conversions, all homozigous Single Nucleotide Polymorphisms (SNP) are extracted while ignoring heterozigous SNPS (hetSNP), Multiple Nucleotide Polymorphisms (MNP) and Insertion/Deletion events (InDel). For each individual, the reference's nucleotide will be assigned unless a SNP is presented. If any individual has a MNP, hetSNP or InDel at a given position, this position is skipped entirely. A General Feature Format (GFF) describing coordinates is used to split the genome into segments. Those segments can be genes, even sized fragments (10kb, 50kb, etc) or particular segments of interest as long as the coordinates are the same as the VCF files. A auxiliary script is provided to generate evenly sized segments. For each selected segment a fasta file will be generated and FastTree will create a distance matrix and a Newick Tree. After all data has been processed, the three files (fasta, matrix and newick) will be read and converted to a database. The webserver scripts will read and serve the data to a web browser. There are three scripts, a main script serves the data and two auxiliary servers to perform on-the-fly clustering and image conversion (from SVG to PNG).
Enter the introgression browser folder
cd ~/introgressionbrowser/
If in a VM, check if your files were correctly shared by virtual box.
ls data
if you don't see your files, there's a mistake in the VM configuration. If you see you data, you can proceed. Enter the data folder. The folder structure should be as follows:
~/introgressionbrowser/ ~/introgressionbrowser/data/ ~/introgressionbrowser/data/analysis/ ~/introgressionbrowser/data/analysis/input/
Inside the data/analysis folder, create a symlink to the executables:
cd data/analysis ln -s ../../vcfmerger .
add all your VCF files inside input folder. add your reference fasta file in the base folder. create a TAB delimited file containing the name of your input files (\t stands for TAB):
1\tinput/file1.vcf.gz\tspecies 1 1\tinput/file2.vcf.gz\tspecies 2 1\tinput/file2.vcf.gz\tspecies 2
the folder structure should resemble:
~/introgressionbrowser/data/analysis/reference.fasta ~/introgressionbrowser/data/analysis/input/file1.vcf.gz ~/introgressionbrowser/data/analysis/input/file2.vcf.gz ~/introgressionbrowser/data/analysis/input/file2.vcf.gz ~/introgressionbrowser/data/analysis/analysis.csv
Now you can run:
vcfmerger/aux/gen_makefile.py
This will generate a makefile for you project (follow one of the examples in the manual) To run the analysis:
make
It will generate a database output:
~/introgressionbrowser/data/analysis/analysis.sqlite
Now create a link to the data folder:
cd ~/introgressionbrowser/data ln -s analysis/analysis.sqlite .
restart ibrowser: If inside a VM, you can restart ibrowser:
~/introgressionbrowser/restart.sh
Or restart the VM:
sudo shutdown -r now
If not inside a VM, you can restart ibrowser:
cd ~/introgressionbrowser/ pgrep -f ibrowser.py | xargs kill pythob ibrowser.py data/
Run vcfmerger/aux/gen_makefile.py to create a makefile for your project
vcfmerger/aux/gen_makefile.py -h
usage: gen_makefile.py [-h] [-i [INLIST]] [-f [INFASTA]] [-s [SIZE]]
[-p [PROJECT]] [-o [OUTFILE]] [-ec EXCLUDED_CHROMS]
[-ic INCLUDED_CHROMS] [-n] [-m] [-np]
[-t [SUB_THREADS]] [-St [SMART_THREADS]] [-SH] [-SI]
[-SS] [-So [SIMPLIFY_OUTPUT]]
[-Coc [CONCAT_CHROMOSOME]]
[-CoI [CONCAT_IGNORE [CONCAT_IGNORE ...]]]
[-Cos [CONCAT_START]] [-Coe [CONCAT_END]]
[-Cot [CONCAT_THREADS]] [-Cor] [-CoR]
[-CoRm [CONCAT_RILMADS]] [-CoRs [CONCAT_RILMINSIM]]
[-CoRg] [-CoRd] [-Ftt [FASTTREE_THREADS]]
[-Ftb [FASTTREE_BOOTSTRAP]] [-Cle [CLUSTER_EXTENSION]]
[-Clt [CLUSTER_THREADS]] [-Clp] [-Cls] [-Cln] [-Clr]
[-Clc] [-Fic [FILTER_CHROMOSOME]] [-Fig [FILTER_GFF]]
[-FiI [FILTER_IGNORE [FILTER_IGNORE ...]]]
[-Fis [FILTER_START]] [-Fie [FILTER_END]] [-Fik] [-Fin]
[-Fiv] [-Fip FILTER_PROTEIN] [-Dbt DB_READ_THREADS]
Create makefile to convert files.
optional arguments:
-h, --help show this help message and exit
-i [INLIST], --input [INLIST], --inlist [INLIST]
input tab separated file
-f [INFASTA], --fasta [INFASTA], --infasta [INFASTA]
input reference fasta. requires split size
-s [SIZE], --size [SIZE]
split size
-p [PROJECT], --proj [PROJECT], --project [PROJECT]
project name
-o [OUTFILE], --out [OUTFILE], --outfile [OUTFILE]
output name [default: makefile]
-ec EXCLUDED_CHROMS, --excluded-chrom EXCLUDED_CHROMS
Do not use the following chromosomes
-ic INCLUDED_CHROMS, --included-chrom INCLUDED_CHROMS
Use EXCLUSIVELY these chromosomes
-n, --dry, --dry-run dry-run
-m, --merge, --cluster_merge
do merged clustering (resource intensive) [default:
no]
-np, --no-pickle do not generate pickle database [default: no]
-t [SUB_THREADS], --sub_threads [SUB_THREADS]
threads of submake to tree building [default: 5]
-St [SMART_THREADS], --smart_threads [SMART_THREADS]
threads of submake to tree building [default: 5]
-SH, --simplify-include-hetero
Do not simplify heterozygous SNPS
-SI, --simplify-include-indel
Do not simplify indel SNPS
-SS, --simplify-include-singleton
Do not simplify single SNPS
-So [SIMPLIFY_OUTPUT], --simplify-output [SIMPLIFY_OUTPUT]
Simplify output file
-Coc [CONCAT_CHROMOSOME], --concat-chrom [CONCAT_CHROMOSOME], --concat-chromosome [CONCAT_CHROMOSOME]
Concat - Chromosome to filter [all]
-CoI [CONCAT_IGNORE [CONCAT_IGNORE ...]], --concat-ignore [CONCAT_IGNORE [CONCAT_IGNORE ...]], --concat-skip [CONCAT_IGNORE [CONCAT_IGNORE ...]]
Concat - Chromosomes to skip
-Cos [CONCAT_START], --concat-start [CONCAT_START]
Concat - Chromosome start position to filter [0]
-Coe [CONCAT_END], --concat-end [CONCAT_END]
Concat - Chromosome end position to filter [-1]
-Cot [CONCAT_THREADS], --concat-threads [CONCAT_THREADS]
Concat - Number of threads [num chromosomes]
-Cor, --concat-noref Concat - Do not print reference [default: true]
-CoR, --concat-RIL Concat - RIL mode: false]
-CoRm [CONCAT_RILMADS], --concat-RIL-mads [CONCAT_RILMADS]
Concat - RIL percentage of Median Absolute Deviation
to use (smaller = more restrictive): 0.25]
-CoRs [CONCAT_RILMINSIM], --concat-RIL-minsim [CONCAT_RILMINSIM]
Concat - RIL percentage of nucleotides identical to
reference to classify as reference: 0.75]
-CoRg, --concat-RIL-greedy
Concat - RIL greedy convert nucleotides to either the
reference sequence or the alternative sequence: false]
-CoRd, --concat-RIL-delete
Concat - RIL delete invalid sequences: false]
-Ftt [FASTTREE_THREADS], --fasttree_threads [FASTTREE_THREADS]
FastTree - number of threads for fasttree
-Ftb [FASTTREE_BOOTSTRAP], --fasttree_bootstrap [FASTTREE_BOOTSTRAP]
FastTree - fasttree bootstrap
-Cle [CLUSTER_EXTENSION], --cluster-ext [CLUSTER_EXTENSION], --cluster-extension [CLUSTER_EXTENSION]
Cluster - [optional] extension to search. [default:
.matrix]
-Clt [CLUSTER_THREADS], --cluster-threads [CLUSTER_THREADS]
Cluster - threads for clustering [default: 5]
-Clp, --cluster-no-png
Cluster - do not export cluster png
-Cls, --cluster-no-svg
Cluster - do not export cluster svg
-Cln, --cluster-no-tree
Cluster - do not export cluster tree. precludes no png
and no svg
-Clr, --cluster-no-rows
Cluster - no rows clustering
-Clc, --cluster-no-cols
Cluster - no column clustering
-Fic [FILTER_CHROMOSOME], --filter-chrom [FILTER_CHROMOSOME], --filter-chromosome [FILTER_CHROMOSOME]
Filter - Chromosome to filter [all]
-Fig [FILTER_GFF], --filter-gff [FILTER_GFF]
Filter - Gff Coordinate file
-FiI [FILTER_IGNORE [FILTER_IGNORE ...]], --filter-ignore [FILTER_IGNORE [FILTER_IGNORE ...]], --filter-skip [FILTER_IGNORE [FILTER_IGNORE ...]]
Filter - Chromosomes to skip
-Fis [FILTER_START], --filter-start [FILTER_START]
Filter - Chromosome start position to filter [0]
-Fie [FILTER_END], --filter-end [FILTER_END]
Filter - Chromosome end position to filter [-1]
-Fik, --filter-knife Filter - Export to separate files
-Fin, --filter-negative
Filter - Invert gff
-Fiv, --filter-verbose
Filter - Verbose
-Fip FILTER_PROTEIN, --filter-prot FILTER_PROTEIN, --filter-protein FILTER_PROTEIN
Filter - Input Fasta File to convert to Protein
-Dbt DB_READ_THREADS, --db-threads DB_READ_THREADS
Db - Number of threads to read raw files
Run MAKE:
makefile -f makefile_[project name]
Copy [project name].sqlite to iBrowser/data folder
cp [project name].sqlite ..
Create [project name].sqlite.nfo with the information about the database:
#title as shall be shown in the UI
title=Tomato 60 RIL - 50k
#custom orders are optional.
#more than one can be given in separate lines
custom_order=RIL.customorder
(OPTIONAL) create custom order files:
#NAME=RIL Single
##NAME is the name of this particular ordering as it will appear in the UI
##
#ROWNUM=1
##ROWNUM is the column to read 1 in the "row order" section
##
##CHROMOSOME=
##CHROMOSOME can either be __global__/empty for ordering all chromosomes, chomosome name for ordering a particular chromosome
##
##row order
ref
S lycopersicum cv MoneyMaker LYC1365
615
634
667
688
710
618
694
678
693
685
651
669
674
676
Reload iBrowser
./vcfmerger/aux/gen_makefile.py --input arabidopsis.csv --infasta TAIR10.fasta --size 50000 --project arabidopsis_50k --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --excluded-chrom chloroplast --excluded-chrom mitochondria --cluster-no-cols make -f makefile_arabidopsis_50k
./vcfmerger/aux/gen_makefile.py --input short2.lst --infasta S_lycopersicum_chromosomes.2.40.fa --size 10000 --project tom84_10k --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --cluster-no-cols make -f makefile_tom84_10k
./vcfmerger/aux/gen_makefile.py --input short2.lst --infasta S_lycopersicum_chromosomes.2.40.fa --size 50000 --project tom84_50k --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --cluster-no-cols make -f makefile_tom84_50k
./vcfmerger/aux/gen_makefile.py --input short2.lst --filter-gff ITAG2.3_gene_models.gff3.gene.gff3 --project tom84_genes --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --cluster-no-cols make -f makefile_tom84_genes
./vcfmerger/aux/gen_makefile.py --input short2.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_10000_introgression.gff --project tom84_10k_introgression --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --cluster-no-cols make -f makefile_tom84_10k_introgression
./vcfmerger/aux/gen_makefile.py --input RIL.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_50000.gff --project RIL_50k --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --cluster-no-cols make -f makefile_RIL_50k
./vcfmerger/aux/gen_makefile.py --input RIL.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_50000.gff --project RIL_50k_mode_ril --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --concat-RIL --cluster-no-cols make -f makefile_RIL_50k_mode_ril
./vcfmerger/aux/gen_makefile.py --input RIL.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_50000.gff --project RIL_50k_mode_ril_greedy --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --concat-RIL --concat-RIL-greedy --cluster-no-cols make -f makefile_RIL_50k_mode_ril_greedy
./vcfmerger/aux/gen_makefile.py --input RIL.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_50000.gff --project RIL_50k_mode_ril_delete --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --concat-RIL --concat-RIL-delete --cluster-no-cols make -f makefile_RIL_50k_mode_ril_delete
./vcfmerger/aux/gen_makefile.py --input RIL.lst --filter-gff S_lycopersicum_chromosomes.2.40.fa_50000.gff --project RIL_50k_mode_ril_delete_greedy --no-pickle --cluster-no-svg --smart_threads 25 --cluster-threads 5 --concat-RIL --concat-RIL-greedy --concat-RIL-delete --cluster-no-cols make -f makefile_RIL_50k_mode_ril_delete_greedy
Merge VCF files:
vcfmerger/vcfmerger.py short.lst
OUTPUT: short.lst.vcf.gz
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT FILENAMES
SL2.40ch00 280 . A C . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S cheesemaniae (055)
SL2.40ch00 284 . A G . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S cheesemaniae (054)
SL2.40ch00 316 . C T . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S arcanum (059)
SL2.40ch00 323 . C T . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S arcanum (059)
SL2.40ch00 332 . A T . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S pimpinellifolium (047)
SL2.40ch00 362 . G T . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S galapagense (104)
SL2.40ch00 385 . A C . PASS NV=1;NW=1;NS=1;NT=1;NU=1 FI S neorickii (056)
SL2.40ch00 391 . C T . PASS NV=1;NW=1;NS=6;NT=6;NU=6 FI S chiemliewskii (052),S neorickii (056),S arcanum (059),S habrochaites glabratum (066),S habrochaites glabratum (067),S habrochaites (072)
Simplify merged VCF deleting hetSNP, MNP and InDels:
vcfmerger/vcfsimplify.py short.lst.vcf.gz
OUTPUT: short.lst.vcf.gz.filtered.vcf.gz
SL2.40ch00 391 . C T . PASS NV=1;NW=1;NS=6;NT=6;NU=6 FI S arcanum (059),S chiemliewskii (052),S habrochaites (072),S habrochaites glabratum (066),S habrochaites glabratum (067),S neorickii (056)
SL2.40ch00 416 . T A . PASS NV=1;NW=1;NS=6;NT=6;NU=6 FI S arcanum (059),S chiemliewskii (052),S habrochaites (072),S habrochaites glabratum (066),S habrochaites glabratum (067),S neorickii (056)
SL2.40ch00 424 . C T . PASS NV=1;NW=1;NS=5;NT=5;NU=5 FI LA0113 (039),S cheesemaniae (054),S pimpinellifolium (044),S pimpinellifolium unc (045),S pimpinellifolium (047)
Generate even sized fragments (if needed):
vcfmerger/aux/fasta_spacer.py GENOME.fa 50000
OUTPUT: GENOME.fa.50000.gff
SL2.40ch00 . fragment_10000 1 10000 . . . Alias=Frag_SL2.40ch00g10000_1;ID=fragment:Frag_SL2.40ch00g10000_1;Name=Frag_SL2.40ch00g10000_1;length=10000;csize=21805821
SL2.40ch00 . fragment_10000 10001 20000 . . . Alias=Frag_SL2.40ch00g10000_2;ID=fragment:Frag_SL2.40ch00g10000_2;Name=Frag_SL2.40ch00g10000_2;length=10000;csize=21805821
Filter with gff:
vcfmerger/vcffiltergff.py -k -f PROJNAME -g GENOME.fa_50000.gff -i short2.lst.vcf.gz.simplified.vcf.gz 2>&1 | tee short2.lst.vcf.gz.simplified.vcf.gz.log
OUTPUT:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT FILENAMES
SL2.40ch00 391 . C T . PASS NV=1;NW=1;NS=6;NT=6;NU=6 FI S arcanum (059),S chiemliewskii (052),S habrochaites (072),S habrochaites glabratum (066),S habrochaites glabratum (067),S neorickii (056)
Concatenate the SNPs of each fragment into FASTA:
find PROJNAME -name '*.vcf.gz' | xargs -I{} -P50 bash -c 'vcfmerger/vcfconcat.py -f -i {} 2>&1 | tee {}.concat.log'
OUTPUT: PROJNAME/CHROMOSOME/short2.lst.vcf.gz.simplified.vcf.gz.filtered.vcf.gz.SL2.40ch01.000090300001-000090310000.Frag_SL2.40ch01g10000_9031.vcf.gz.SL2.40ch01.fasta
>Moneymaker_001
ATAATCTAGCTGGAACCCTTGTTTTTCTCGCGATTGGGGTTCAAGTGCACACCACATGTC
AGGGA
>Alisa_Craig_002
ATAATCTAGCTGGAACCCTTGTTTTTCTTGCGATTGGGGTTCAAGTGCGCGCTGCGTGAC
AGGAA
Run FastTree in each of the FASTA files:
export OMP_NUM_THREADS=3
find PROJNAME -name '*.fasta' | sort | xargs -I{} -P30 bash -c 'vcfmerger/aux/FastTreeMP -fastest -gamma -nt -bionj -boot 100 -log {}.tree.log -out {}.tree {}'
OUTPUT: PROJNAME/CHROMOSOME/short2.lst.vcf.gz.simplified.vcf.gz.filtered.vcf.gz.SL2.40ch01.000090300001-000090310000.Frag_SL2.40ch01g10000_9031.vcf.gz.SL2.40ch01.fasta.tree
((((Dana_018:0.0,Belmonte_033:0.0):0.00054,((TR00026_102:0.01587,(PI272654_023:0.03426,(((S_huaylasense_063:0.00054,((Lycopersicon_sp_025:0.0,S_chilense_065:0.0):0.00054,S_chilense_064:0.01555)0.780:0.01548)0.860:0.01547,((S_peruvianum_new_049:0.0,S_chiemliewskii_051:0.0,S_chiemliewskii_052:0.0,S_cheesemaniae_053:0.0,S_cheesemaniae_054:0.0,S_neorickii_056:0.0,S_neorickii_057:0.0,S_peruvianum_060:0.0,S_habrochaites_glabratum_066:0.0,S_habrochaites_glabratum_068:0.0,S_habrochaites_070:0.0,S_habrochaites_071:0.0,S_habrochaites_072:0.0,S_pennellii_073:0.0,S_pennellii_074:0.0,TR00028_LA1479_105:0.0,ref:0.0):0.00054,((S_arcanum_058:0.01482,(S_huaylasense_062:0.08258,S._arcanum_new_075:0.00054)0.880:0.03260)0.960:0.04917,(((Gardeners_Delight_003:0.00054,(Katinka_Cherry_007:0.0,Trote_Beere_016:0.0,Winter_Tipe_031:0.0):0.01559)0.900:0.03206,(PI129097_022:0.00054,(S_galapagense_104:0.04782,(LA0113_039:0.01223,((S_pimpinellifolium_047:0.01628,(S_arcanum_059:0.00055,(S_habrochaites_glabratum_067:0.01562,S_habrochaites_glabratum_069:0.01562)1.000:0.08287)0.920:0.04857)0.670:0.01186,S_habrochaites_042:0.03551)0.990:0.12956)0.960:0.06961)0.710:0.00054)0.800:0.01578)0.760:0.01558,(T1039_017:0.08246,S_pimpinellifolium_044:0.00054)0.980:0.08153)0.230:0.00053)0.910:0.00055)0.910:0.00054)0.830:0.01549,S_pimpinellifolium_046:0.00054)0.980:0.08610)0.660:0.01369)0.530:0.04644,(TR00027_103:0.00054,(PI365925_037:0.04936,S_cheesemaniae_055:0.03179)0.650:0.08462)1.000:0.41706)0.650:0.00296)0.940:0.01555,(The_Dutchman_028:0.00053,(((Polish_Joe_026:0.0,Brandywine_089:0.0):0.00054,((((Porter_078:0.01608,Kentucky_Beefsteak_093:0.01542)0.880:0.03271,(Thessaloniki_096:0.08543,Bloodt_Butcher_088:0.03267)0.700:0.01564)0.800:0.01585,(Giant_Belgium_091:0.01562,(Moneymaker_001:0.00054,(Dixy_Golden_Giant_090:0.01579,(Large_Red_Cherry_077:0.03276,Momatero_015:0.04969)0.720:0.01528)0.870:0.01570)0.850:0.01556)0.480:0.00055)0.930:0.03157,Marmande_VFA_094:0.03158)0.970:0.00053)0.880:0.00053,Watermelon_Beefsteak_097:0.01555)0.890:0.01559)0.970:0.03159)0.950:0.00054,PI169588_041:0.00054,((Sonato_012:0.11798,(((All_Round_011:0.01555,Chih-Mu-Tao-Se_038:0.00054)0.180:0.00054,(((Jersey_Devil_024:0.0,Chag_Li_Lycopersicon_esculentum_032:0.0,S_pimpinellifolium_unc_043:0.0):0.00054,(((PI311117_036:0.04839,((Taxi_006:0.0,Tiffen_Mennonite_034:0.0):0.00054,(Cal_J_TM_VF_027:0.00053,(Lycopersicon_esculentum_828_021:0.00054,(Black_Cherry_029:0.03245,(Galina_005:0.00054,S_pimpinellifolium_unc_045:0.01559)0.880:0.03248)0.770:0.01547)0.950:0.03179)0.160:0.01560)0.840:0.01563)0.420:0.00054,Lycopersicon_esculentum_825_020:0.00054)0.860:0.01556,((Cross_Country_013:0.0,ES_58_Heinz_040:0.0):0.00054,(Rutgers_004:0.01554,Lidi_014:0.04758)0.900:0.00054)0.880:0.00054)0.860:0.01558)0.080:0.01560,(Alisa_Craig_002:0.01560,John_s_big_orange_008:0.00054)1.000:0.00054)0.840:0.01558)0.800:0.01566,(Large_Pink_019:0.01555,Anto_030:0.00054)0.140:0.00054)0.920:0.01555)0.680:0.00054,Wheatley_s_Frost_Resistant_035:0.03155)0.950:0.00054);
find PROJNAME -name '*.fasta' | sort | xargs -I{} -P30 bash -c 'vcfmerger/aux/FastTreeMP -nt -makematrix {} > {}.matrix'
OUTPUT: PROJNAME/CHROMOSOME/short2.lst.vcf.gz.simplified.vcf.gz.filtered.vcf.gz.SL2.40ch01.000090300001-000090310000.Frag_SL2.40ch01g10000_9031.vcf.gz.SL2.40ch01.fasta.matrix
Moneymaker_001 0.000000 0.134437 0.345611 0.134437 0.321609
Alisa_Craig_002 0.134437 0.000000 0.211925 0.064210
Gardeners_Delight_003 0.345611 0.211925 0.000000 0.211925
Process the data into memory dump database (pyckle):
vcf_walk_ram.py --pickle PROJNAME
OUTPUT:
walk_out_10k.db
walk_out_10k_SL2.40ch00.db
walk_out_10k_SL2.40ch01.db
walk_out_10k_SL2.40ch02.db
walk_out_10k_SL2.40ch03.db
walk_out_10k_SL2.40ch04.db
walk_out_10k_SL2.40ch05.db
walk_out_10k_SL2.40ch06.db
walk_out_10k_SL2.40ch07.db
walk_out_10k_SL2.40ch08.db
walk_out_10k_SL2.40ch09.db
walk_out_10k_SL2.40ch10.db
walk_out_10k_SL2.40ch11.db
walk_out_10k_SL2.40ch12.db
Convert (pickle) database to SQLite (if dependencies installed):
vcf_walk_sql.py PROJNAME
OUTPUT:
walk_out_10k.sqlite
This project is maintained by Saulo Aflitos ( GitHub and LinkedIn ) with support from Applied Bioinformatics and WageningenUR
Hosted on GitHub Pages — Theme by orderedlist
<script type="text/javascript">var gaJsHost = (("https:" == document.location.protocol) ? "https://ssl." : "http://www."); document.write(unescape("%3Cscript src='" + gaJsHost + "google-analytics.com/ga.js' type='text/javascript'%3E%3C/script%3E"));</script> <script type="text/javascript">try { var pageTracker = _gat._getTracker("UA-5291039-9"); pageTracker._trackPageview(); } catch(err) {}</script>