Start gwas.sif container, generate synthetic phenotype with 100 causal genetic variants, and run plink2 to compute GWAS p-values.
This is a demo using synthetic genotypes (10K subjects, 150K SNPs), produced by HapGen2 tool.
singularity shell --no-home -B $COMORMENT_REF:/ref -B data:/data gwas.sif
simu_linux --bfile /ref/hapgen/chr21 --qt --causal-n 100 --out /data/simu
plink2 --bfile /ref/hapgen/chr21 --glm allow-no-covars --pheno /data/simu.pheno --pheno-name trait1 --out /data/plink
Start python3.sif container, and generate QQ plot and Manhattan plots.
singularity shell --no-home -B $COMORMENT_REF:/ref -B data:/data python3.sif
python /tools/python_convert/qq.py /data/plink.trait1.glm.linear --p P --snp ID --out /data/plink.trait1.glm.linear.qq.png
python /tools/python_convert/manhattan.py /data/plink.trait1.glm.linear --p P --snp ID --bp POS --chr "#CHROM" --out /data/plink.trait1.glm.linear.manh
Use PRSice to compute polygenic risk scores on synthetic dataset.
Start gwas.sif container, and execute PRSice_linux tool:
singularity shell --no-home -B $COMORMENT_REF:/ref -B data:/data gwas.sif
/tools/prsice/PRSice_linux \
--base /data/plink.trait1.glm.linear --stat BETA --beta --chr "#CHROM" --bp POS --snp ID --pvalue P \
--target /ref/hapgen/chr21 \
--pheno /data/simu.pheno --pheno-col trait1 \
--fastscore --bar-levels 5e-08,1e-6,1e-5,1e-4,1e-3,0.01,0.05,0.1,0.5,1 \
--out /data/trait1
Result in trait1.prsice file:
Pheno Set Threshold R2 P Coefficient Standard.Error Num_SNP
- Base 5e-08 0.551604 0 110.673 0.997937 103
- Base 1e-06 0.561591 0 148.262 1.31009 153
- Base 1e-05 0.563446 0 182.494 1.60652 206
- Base 0.0001 0.568096 0 237.958 2.07504 300
- Base 0.001 0.584015 0 348.353 2.94029 505
- Base 0.01 0.616787 0 623.339 4.91383 1108
- Base 0.05 0.651179 0 1079.49 7.90152 2317
- Base 0.1 0.666187 0 1458.03 10.322 3463
- Base 0.5 0.688963 0 3103.9 20.8574 8735
- Base 1 0.690659 0 4249.09 28.4397 12101
Start ldsc.sif container:
singularity shell --no-home -B $COMORMENT_REF:/ref -B data:/data ldsc.sif
Pre-process summary statistics with munge_sumstats.py script:
TBD: investigate why these commands were so slow...
python /tools/ldsc/munge_sumstats.py --merge-alleles /ref/ldsc/w_hm3.snplist --sumstats /ref/sumstats/PGC_BIP_2016.sumstats.gz --out /data/PGC_BIP_2016 --signed-sumstats Z,0
mv /data/PGC_BIP_2016.sumstats.gz /data/PGC_BIP_2016.sumstats && gzip /data/PGC_BIP_2016.sumstats # hack-hack, ldsc produces .gz files which aren't actually compressed
python /tools/ldsc/munge_sumstats.py --merge-alleles /ref/ldsc/w_hm3.snplist --sumstats /ref/sumstats/PGC_SCZ_2014_EUR.sumstats.gz --out /data/PGC_SCZ_2014_EUR --signed-sumstats Z,0
mv /data/PGC_SCZ_2014_EUR.sumstats.gz /data/PGC_SCZ_2014_EUR.sumstats && gzip /data/PGC_SCZ_2014_EUR.sumstats
Calculate genetic correlation:
python /tools/ldsc/ldsc.py --rg /data/PGC_SCZ_2014_EUR.sumstats.gz,/data/PGC_BIP_2016.sumstats.gz \
--ref-ld-chr /ref/ldsc/eur_w_ld_chr/ \
--w-ld-chr /ref/ldsc/eur_w_ld_chr/ \
--out /data/ldsc_rg
Result:
Summary of Genetic Correlation Results
p1 p2 rg se z p h2_obs h2_obs_se h2_int h2_int_se gcov_int gcov_int_se
/data/PGC_SCZ_2014_EUR.sumstats.gz /data/PGC_BIP_2016.sumstats.gz 0.6815 0.0211 32.2318 6.3311e-228 0.3476 0.0175 1.0201 0.0102 0.244 0.0085
singularity shell --no-home -B $COMORMENT_REF:/ref -B data:/data python3.sif
python /tools/python_convert/sumstats.py clump \
--sumstats /ref/sumstats/PGC_SCZ_2014_EUR.sumstats.gz \
--out /data/PGC_SCZ_2014_EUR.clump \
--bfile-chr /ref/1000genomes/[email protected]
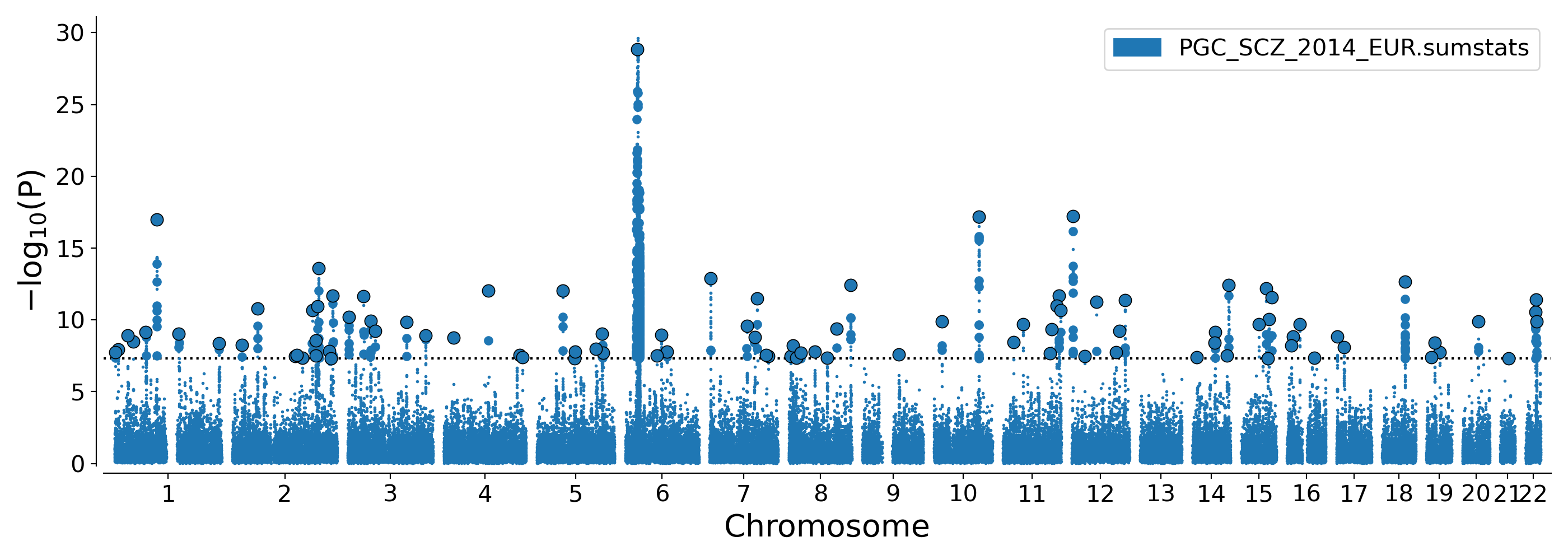
python /tools/python_convert/manhattan.py /ref/sumstats/PGC_SCZ_2014_EUR.sumstats.gz \
--lead /data/PGC_SCZ_2014_EUR.clump.lead.csv \
--indep /data/PGC_SCZ_2014_EUR.clump.indep.csv \
--out /data/PGC_SCZ_2014_EUR.manh
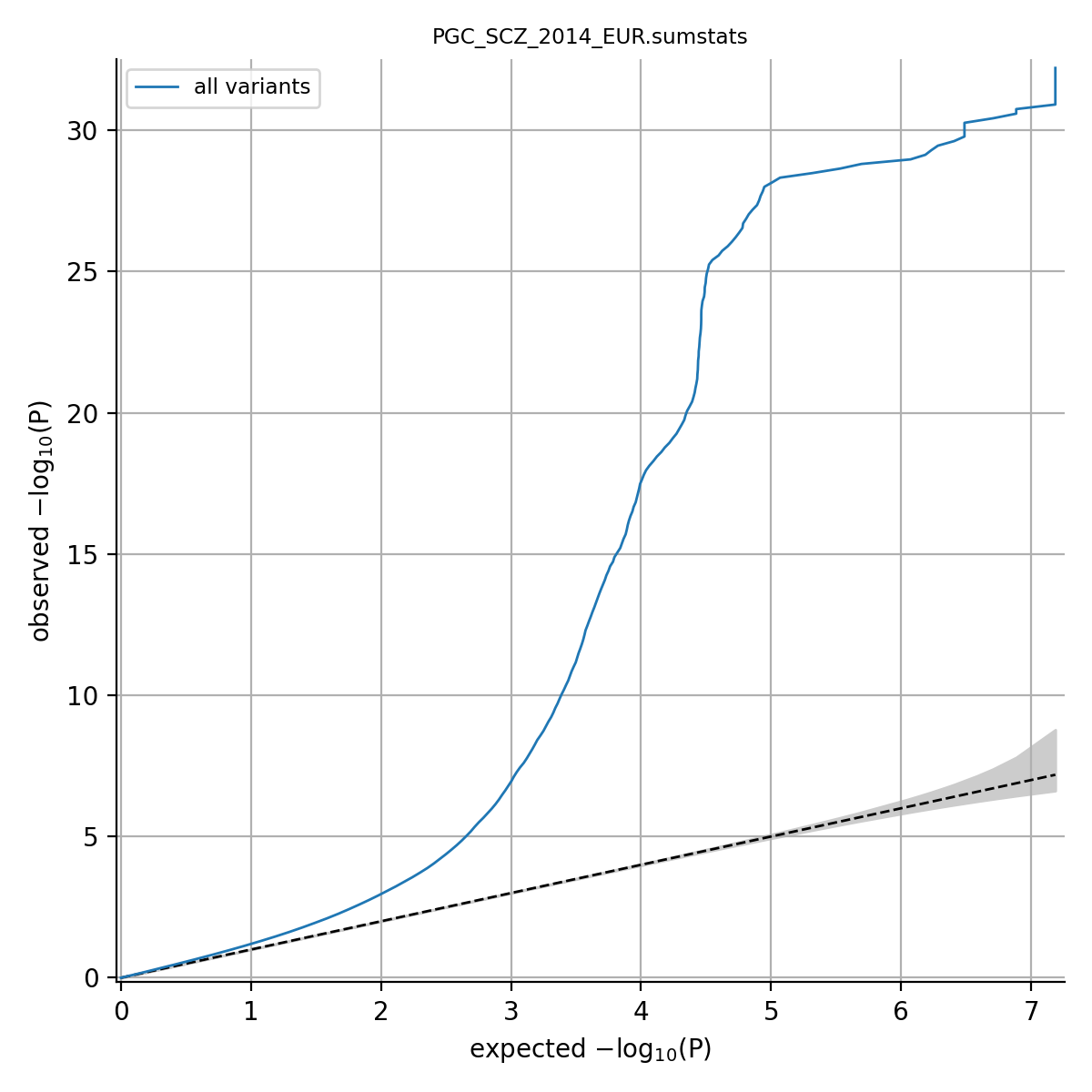
python /tools/python_convert/qq.py /ref/sumstats/PGC_SCZ_2014_EUR.sumstats.gz --out /data/PGC_SCZ_2014_EUR.qq.png
Result:
$head PGC_SCZ_2014_EUR.clump.loci.csv
locusnum CHR LEAD_SNP LEAD_BP MinBP MaxBP PVAL
1 1 rs4648845 2387101 2369498 2402499 1.737e-08
2 1 rs301797 8487323 8404093 8701288 1.202e-08
3 1 rs1498232 30433951 30412503 30437118 1.206e-09
4 1 rs11210892 44100084 44029353 44137257 3.417e-09
5 1 rs35998080 73278615 73275828 74033340 6.953e-10
...