Yujia Li, Oriol Vinyals, Chris Dyer, Razvan Pascanu, and Peter Battaglia. Learning Deep Generative Models of Graphs. arXiv preprint arXiv:1803.03324, 2018.
DGMG generates graphs by progressively adding nodes and edges as below:
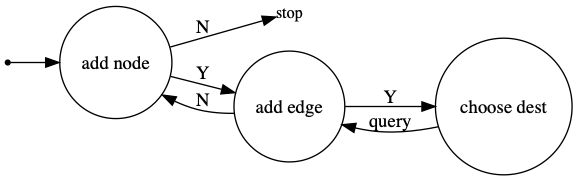
For molecules, the nodes are atoms and the edges are bonds.
Goal: Given a set of real molecules, we want to learn the distribution of them and get new molecules
with similar properties. See the Evaluation section for more details.
With our implementation, this model has several limitations:
- Information about protonation and chirality are ignored during generation
- Molecules consisting of
[N+],[O-], etc. cannot be generated.
For example, the model can only generate O=C1NC(=S)NC(=O)C1=CNC1=CC=C(N(=O)O)C=C1O from
O=C1NC(=S)NC(=O)C1=CNC1=CC=C([N+](=O)[O-])C=C1O even with the correct decisions.
To avoid issues about validity and novelty, we filter out these molecules from the dataset.
The authors use the ChEMBL database. Since they did not release the code, we use a subset from Olivecrona et al., another work on generative modeling.
The authors restrict their dataset to molecules with at most 20 heavy atoms, and used a training/validation split of 130, 830/26, 166 examples each. We use the same split but need to relax 20 to 23 as we are using a different subset.
After the pre-processing, we are left with 232464 molecules for training and 5000 molecules for validation.
Training auto-regressive generative models tends to be very slow. According to the authors, they use multiprocess to speed up training and gpu does not give much speed advantage. We follow their approach and perform multiprocess cpu training.
To start training, use train.py with required arguments
-d DATASET, dataset to use (default: None), built-in support exists for ChEMBL, ZINC
-o {random,canonical}, order to generate graphs (default: None)
and optional arguments
-s SEED, random seed (default: 0)
-np NUM_PROCESSES, number of processes to use (default: 32)
Even though multiprocess yields a significant speedup comparing to a single process, the training can still take a long time (several days). An epoch of training and validation can take up to one hour and a half on our machine. If not necessary, we recommend users use our pre-trained models.
Meanwhile, we make a checkpoint of our model whenever there is a performance improvement on the validation set so you do not need to wait until the training terminates.
All training results can be found in training_results.
You can also use your own dataset with additional arguments
-tf TRAIN_FILE, Path to a file with one SMILES a line for training
data. This is only necessary if you want to use a new
dataset. (default: None)
-vf VAL_FILE, Path to a file with one SMILES a line for validation
data. This is only necessary if you want to use a new
dataset. (default: None)
We can monitor the training process with tensorboard as below:

To use tensorboard, you need to install tensorboardX and
TensorFlow. You can lunch tensorboard with tensorboard --logdir=.
If you are training on a remote server, you can still use it with:
- Launch it on the remote server with
tensorboard --logdir=. --port=A - In the terminal of your local machine, type
ssh -NfL localhost:B:localhost:A username@your_remote_host_name - Go to the address
localhost:Bin your browser
To start evaluation, use eval.py with required arguments
-d DATASET, dataset to use (default: None), built-in support exists for ChEMBL, ZINC
-o {random,canonical}, order to generate graphs, used for naming evaluation directory (default: None)
-p MODEL_PATH, path to saved model (default: None). This is not needed if you want to use pretrained models.
-pr, Whether to use a pre-trained model (default: False)
and optional arguments
-s SEED, random seed (default: 0)
-ns NUM_SAMPLES, Number of molecules to generate (default: 100000)
-mn MAX_NUM_STEPS, Max number of steps allowed in generated molecules to
ensure termination (default: 400)
-np NUM_PROCESSES, number of processes to use (default: 32)
-gt GENERATION_TIME, max time (seconds) allowed for generation with
multiprocess (default: 600)
All evaluation results can be found in eval_results.
After the evaluation, 100000 molecules will be generated and stored in generated_smiles.txt under eval_results
directory, with three statistics logged in generation_stats.txt under eval_results:
Validity among allgives the percentage of molecules that are validUniqueness among valid onesgives the percentage of valid molecules that are uniqueNovelty among unique onesgives the percentage of unique valid molecules that are novel (not seen in training data)
We also provide a jupyter notebook where you can visualize the generated molecules

and compare their property distributions against the training molecule property distributions

You can download the notebook with wget https://data.dgl.ai/dgllife/dgmg/eval_jupyter.ipynb.
Below gives the statistics of pre-trained models. With random order, the training becomes significantly more difficult
as we now have N^2 data points with N molecules.
| Pre-trained model | % valid | % unique among valid | % novel among unique |
|---|---|---|---|
ChEMBL_canonical |
78.80 | 99.19 | 98.60 |
ChEMBL_random |
29.09 | 99.87 | 100.00 |
ZINC_canonical |
74.60 | 99.87 | 99.87 |
ZINC_random |
12.37 | 99.38 | 100.00 |