#安装bowtie(在python<=2.7)
conda install bowtie
#bowtie建立索引(酿酒酵母Saccharomyces cerevisiae、大肠杆菌Escherichia coli)
bowtie-build ./S288C_reference_sequence_R64-3-1_20210421.fsa ./BowtieIndex/YeastGenome
bowtie-build NC_008253.fna ./BowtieIndex/e_coli
#mapping
bowtie -v 2 -m 10 --best --strata BowtieIndex/YeastGenome -f THA1.fa -S THA1.sam
bowtie -v 1 -m 10 --best --strata BowtieIndex/e_coli -q e_coli_1000_1.fq -S e_coli_1000_1.sam
#mapping(不用bowtie-build,用bowtie官网下载的ebwt)
bowtie -v 2 -m 10 --best --strata BowtieIndex/s_cerevisiae.ebwt/s_cerevisiae -f THA1.fa -S THA1.sam
bowtie -v 1 -m 10 --best --strata BowtieIndex/e_coli.ebwt/e_coli -q e_coli_1000_1.fq -S e_coli_1000_1.sam#安装STAR
conda install star
#为索引建立文件夹
mkdir tair10.Pt.STARindex
#STAR建立索引
STAR --runMode genomeGenerate --genomeFastaFiles data/tair10.Pt.fa --sjdbGTFfile data/tair10.Pt.gtf --genomeDir tair10.Pt.STARindex --genomeSAindexNbases 71.请阐述bowtie中利用了 BWT 的什么性质提高了运算速度?并通过哪些策略优化了对内存的需求?
Burrows-Wheeler Transform (BWT) 是文本中字符的可逆排列。虽然最初是在数据压缩的背景下开发的,但基于 BWT 的索引允许在较小的内存占用范围内有效地搜索大文本。该矩阵有一个称为“last first (LF) mapping”的属性。最后一列中第 i次出现的字符 X 与第一列中第 i次出现的 X对应的文本字符相同。此属性是使用 BWT 索引导航或搜索文本的算法的基础。
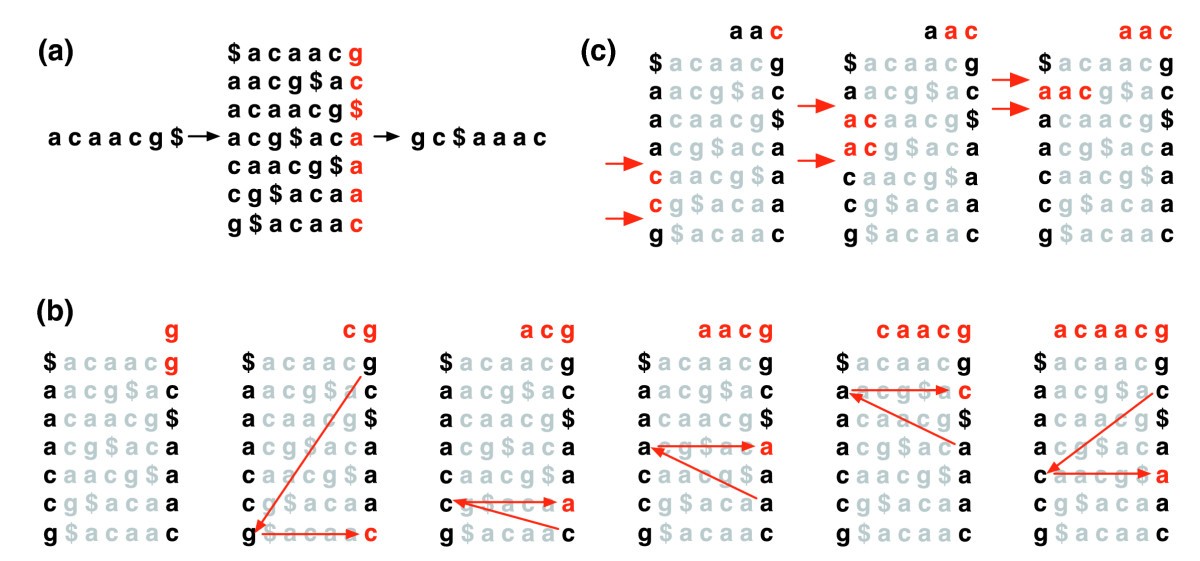
(Searching for inexact alignments) Bowtie引入了一种对齐算法,该算法进行回溯搜索以快速找到满足指定对齐策略的对齐。读数中的每个字符都有一个数字质量值,值越低表示测序错误的可能性越高。bowtie的对齐策略允许有限数量的不匹配,并且更喜欢所有不匹配位置的质量值总和较低的对齐。
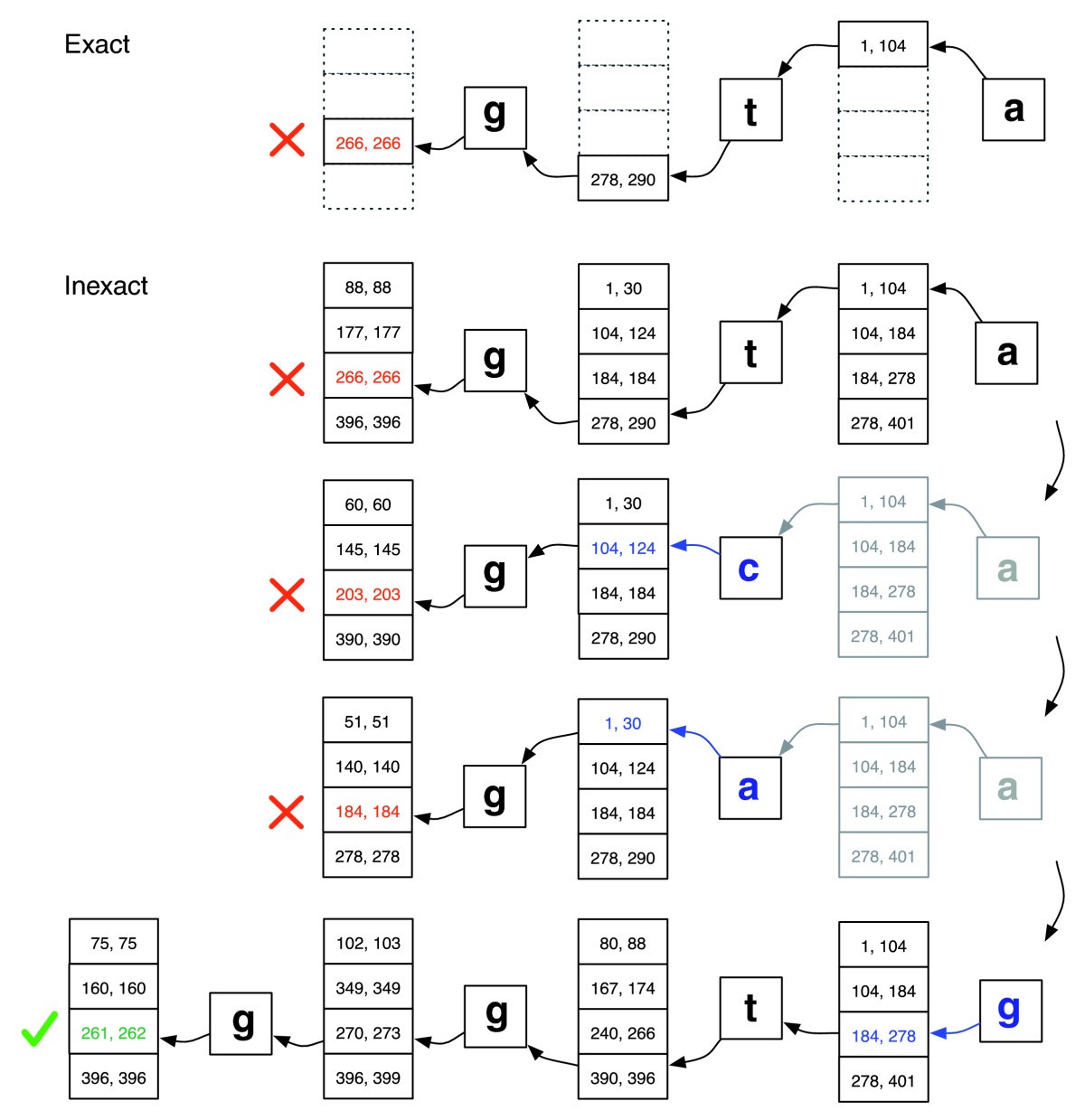
(Excessive backtracking) Bowtie 还通过“双索引”技术减少了过度回溯。创建基因组的两个索引:第一个包含基因组的 BWT,称为“正向”索引,第二个包含基因组的 BWT,其字符序列反转(不是反向互补),称为“镜像”索引。
参考: https://genomebiology.biomedcentral.com/articles/10.1186/gb-2009-10-3-r25 (Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.)
https://www.bilibili.com/video/BV1ix41177d4/
https://www.youtube.com/watch?v=4n7NPk5lwbI
2.用bowtie将 THA2.fa mapping 到 BowtieIndex/YeastGenome 上,得到 THA2.sam,统计mapping到不同染色体上的reads数量(即统计每条染色体都map上了多少条reads)。
#方法一(自行建立索引)
#安装bowtie(在python<=2.7)
conda install bowtie
#bowtie建立索引(酿酒酵母Saccharomyces cerevisiae、大肠杆菌Escherichia coli)
bowtie-build ./S288C_reference_sequence_R64-3-1_20210421.fsa ./BowtieIndex/YeastGenome
#mapping
bowtie -v 2 -m 10 --best --strata BowtieIndex/YeastGenome -f THA2.fa -S THA2.sam
# reads processed: 1250
# reads with at least one reported alignment: 1158 (92.64%)
# reads that failed to align: 77 (6.16%)
# reads with alignments suppressed due to -m: 15 (1.20%)
#Reported 1158 alignments to 1 output stream(s)
#统计染色体上mapping上的reads数(这里显示的是参考基因NC_编号)
grep -v '^@' THA2.sam | cut -f 3 | sort | uniq -c
# 92 *
# 18 ref|NC_001133|
# 51 ref|NC_001134|
# 15 ref|NC_001135|
# 194 ref|NC_001136|
# 33 ref|NC_001137|
# 17 ref|NC_001138|
# 125 ref|NC_001139|
# 68 ref|NC_001140|
# 25 ref|NC_001141|
# 71 ref|NC_001142|
# 56 ref|NC_001143|
# 169 ref|NC_001144|
# 67 ref|NC_001145|
# 58 ref|NC_001146|
# 101 ref|NC_001147|
# 78 ref|NC_001148|
# 12 ref|NC_001224|#方法二(直接用ebwt文件索引)
#安装bowtie(在python<=2.7)
conda install bowtie
#mapping
bowtie -v 2 -m 10 --best --strata BowtieIndex/s_cerevisiae.ebwt/s_cerevisiae -f THA2.fa -S THA2.sam
# reads processed: 1250
# reads with at least one reported alignment: 1158 (92.64%)
# reads that failed to align: 77 (6.16%)
# reads with alignments suppressed due to -m: 15 (1.20%)
#Reported 1158 alignments to 1 output stream(s)
#统计染色体上mapping上的reads数
grep -v '^@' THA2.sam | cut -f 3 | sort | uniq -c
# 92 *
# 18 Scchr01
# 51 Scchr02
# 15 Scchr03
# 194 Scchr04
# 33 Scchr05
# 17 Scchr06
# 125 Scchr07
# 68 Scchr08
# 25 Scchr09
# 71 Scchr10
# 56 Scchr11
# 169 Scchr12
# 67 Scchr13
# 58 Scchr14
# 101 Scchr15
# 78 Scchr16
# 12 Scmito3.查阅资料,回答以下问题: 3.1什么是sam/bam文件中的"CIGAR string"? 它包含了什么信息?
3.2"soft clip"的含义是什么,在CIGAR string中如何表示?
3.3什么是reads的mapping quality? 它反映了什么样的信息?
3.4仅根据sam/bam文件的信息,能否推断出read mapping到的区域对应的参考基因组序列?
提示:参考https://samtools.github.io/hts-specs/SAMtags.pdf中对于MD tag的介绍
4.软件安装和资源文件的下载也是生物信息学实践中的重要步骤。请自行安装教程中未涉及的 bwa 软件,从UCSC Genome Browser下载Yeast (S. cerevisiae, sacCer3)基因组序列。使用bwa对Yeast基因组sacCer3.fa建立索引,并利用bwa将THA2.fa,mapping到Yeast参考基因组上,并进一步转化输出得到THA2-bwa.sam文件。